
Combined Transcriptomic and Metabolomic Analysis Reveals the Mechanism of Flavonoid Biosynthesis in Handroanthus chrysanthus (Jacq.) S.O.Grose


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. Metabolite Extraction
2.3. LC-MS/MS Analysis and Data Processing
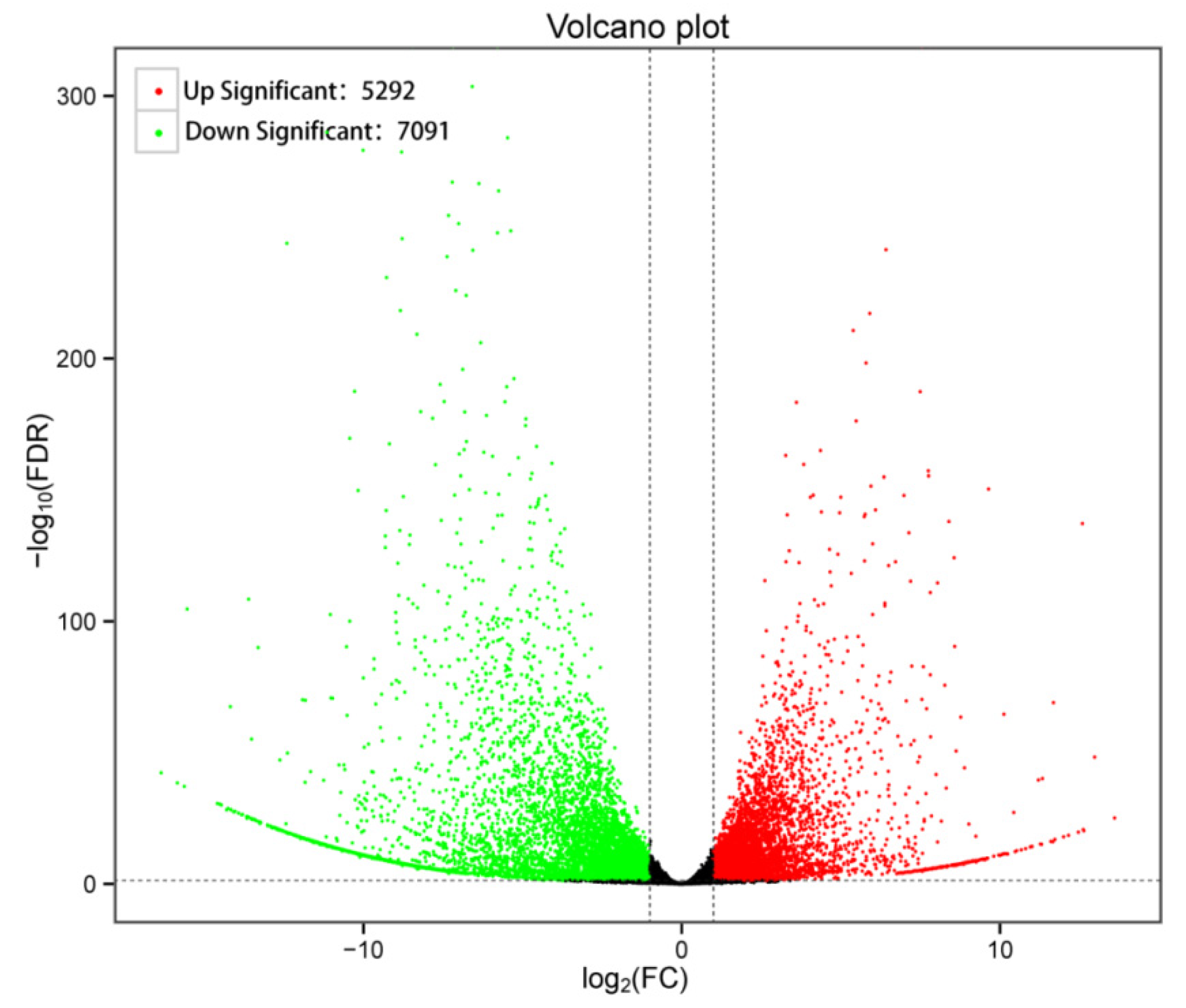
2.4. High Throughput Sequencing for Transcriptomic Analysis and Data Processing
2.5. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.6. Combined Transcriptomic–Metabolomic Analysis
3. Results
3.1. Multivariate Statistical Analysis
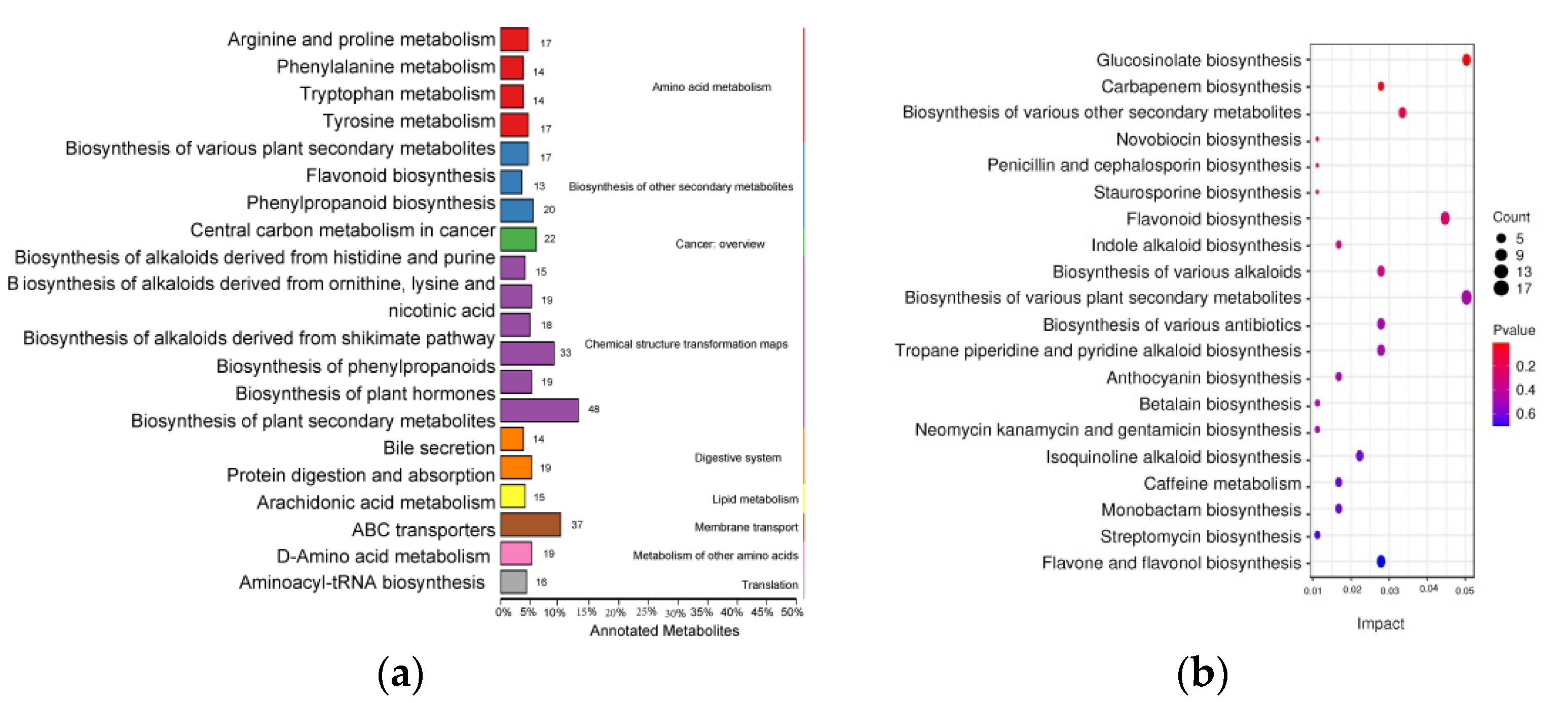
3.2. Metabolites in the Bark and Leaves of H. chrysantha
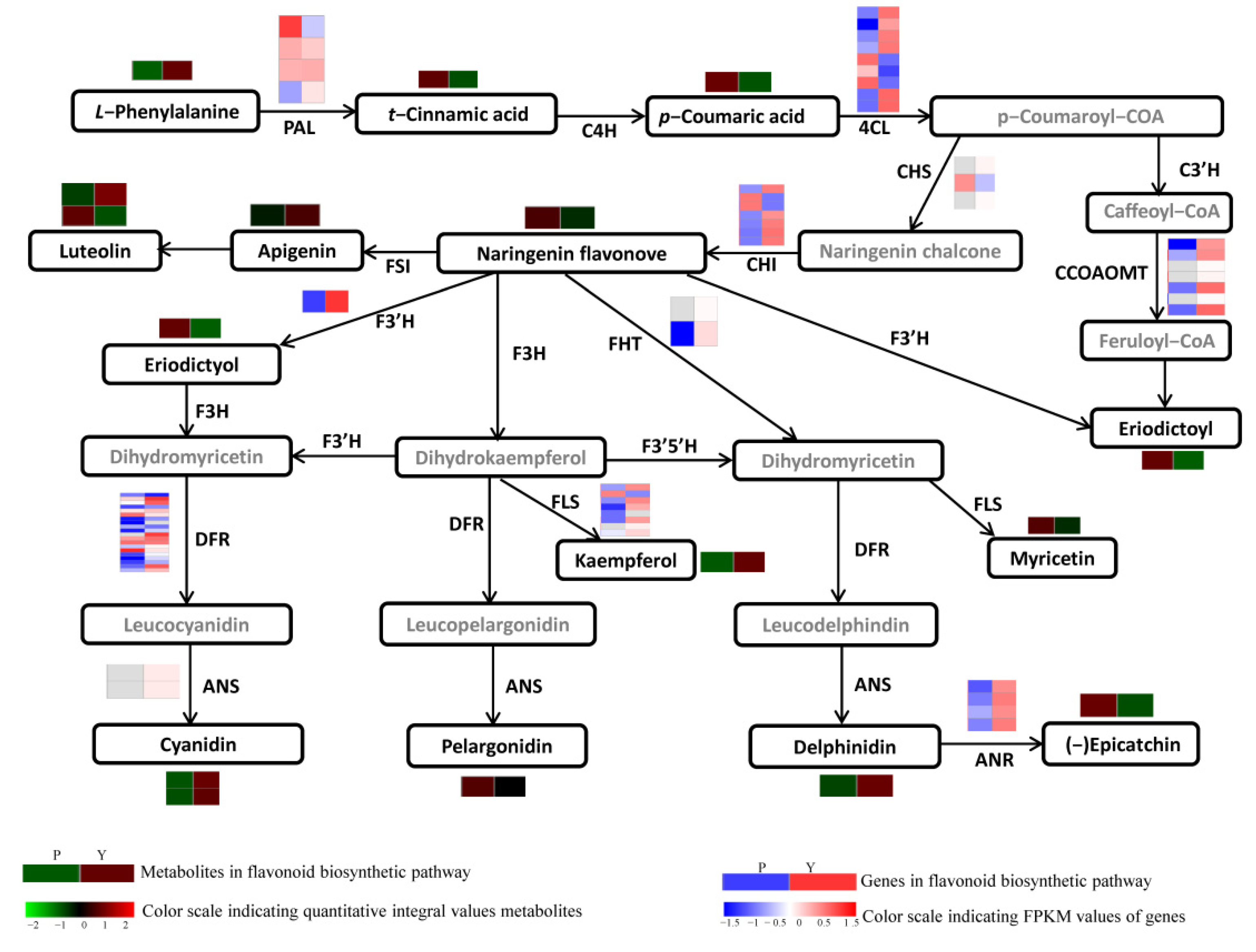
3.3. Secondary Metabolites Identified in the Flavonoid Pathway
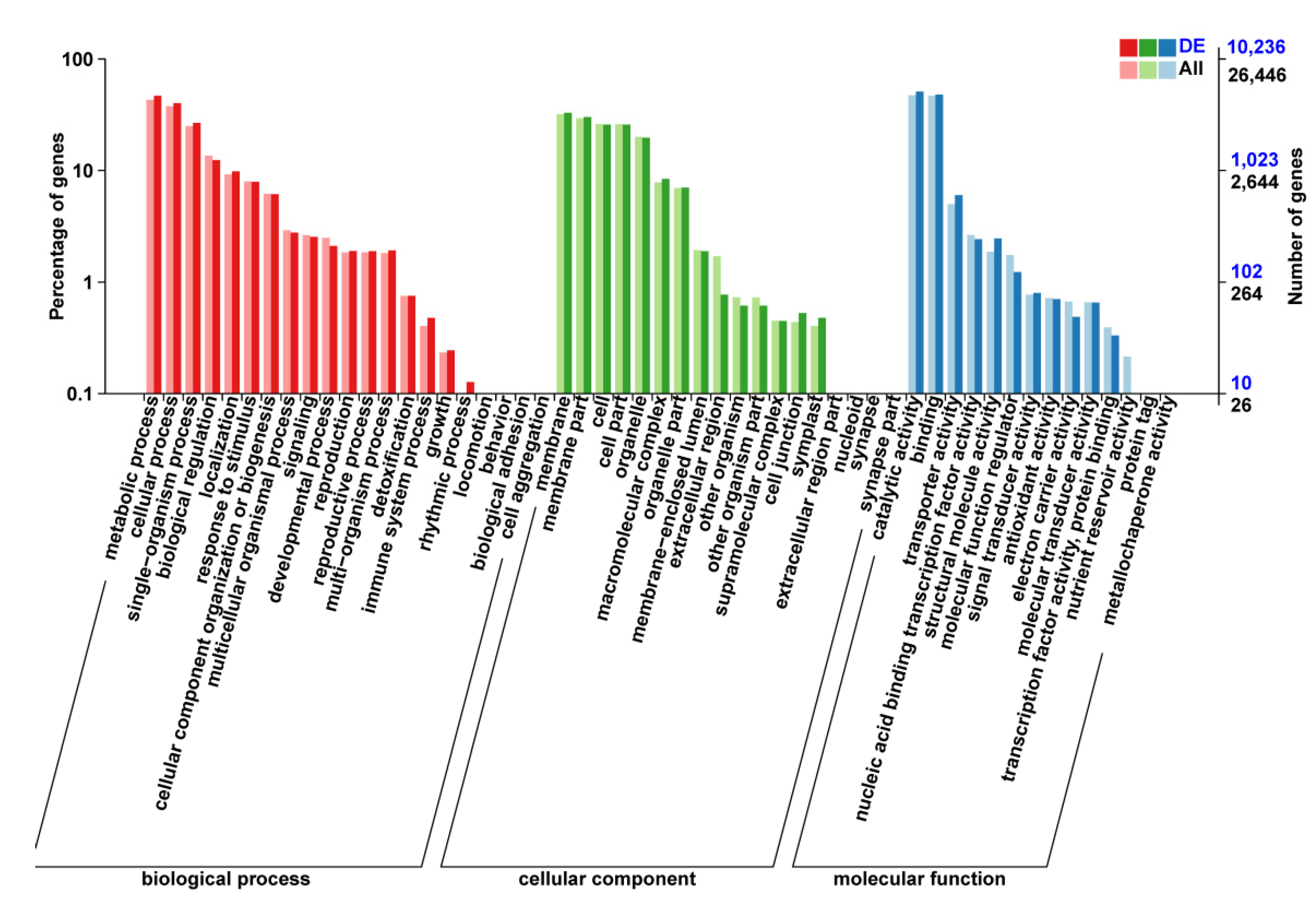
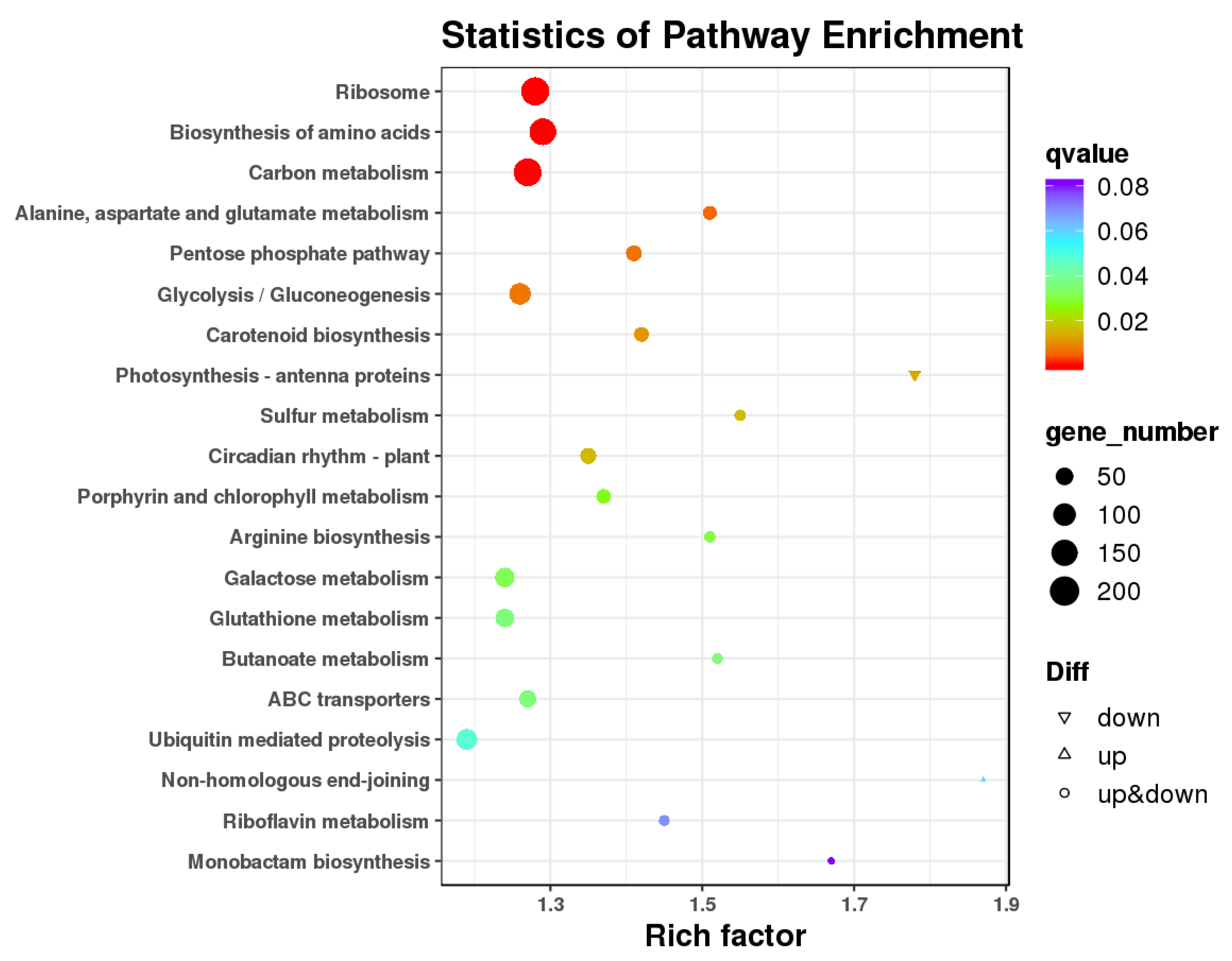
3.4. GO Enrichment and KEGG Pathway Analysis of DEGs
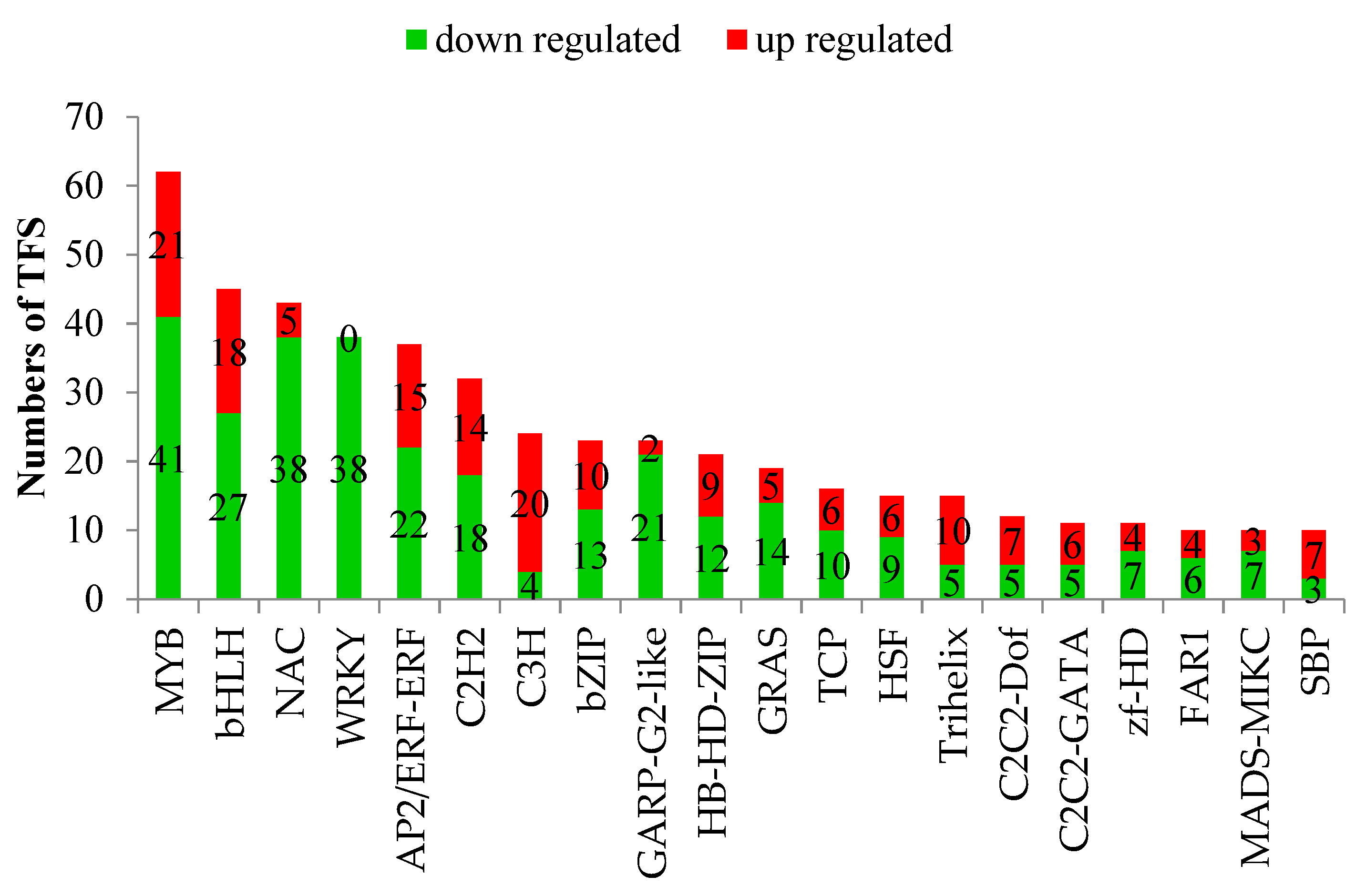
3.5. Analysis of Transcription Factors (TF)
3.6. Enzyme Genes in Flavonoid Biosynthesis
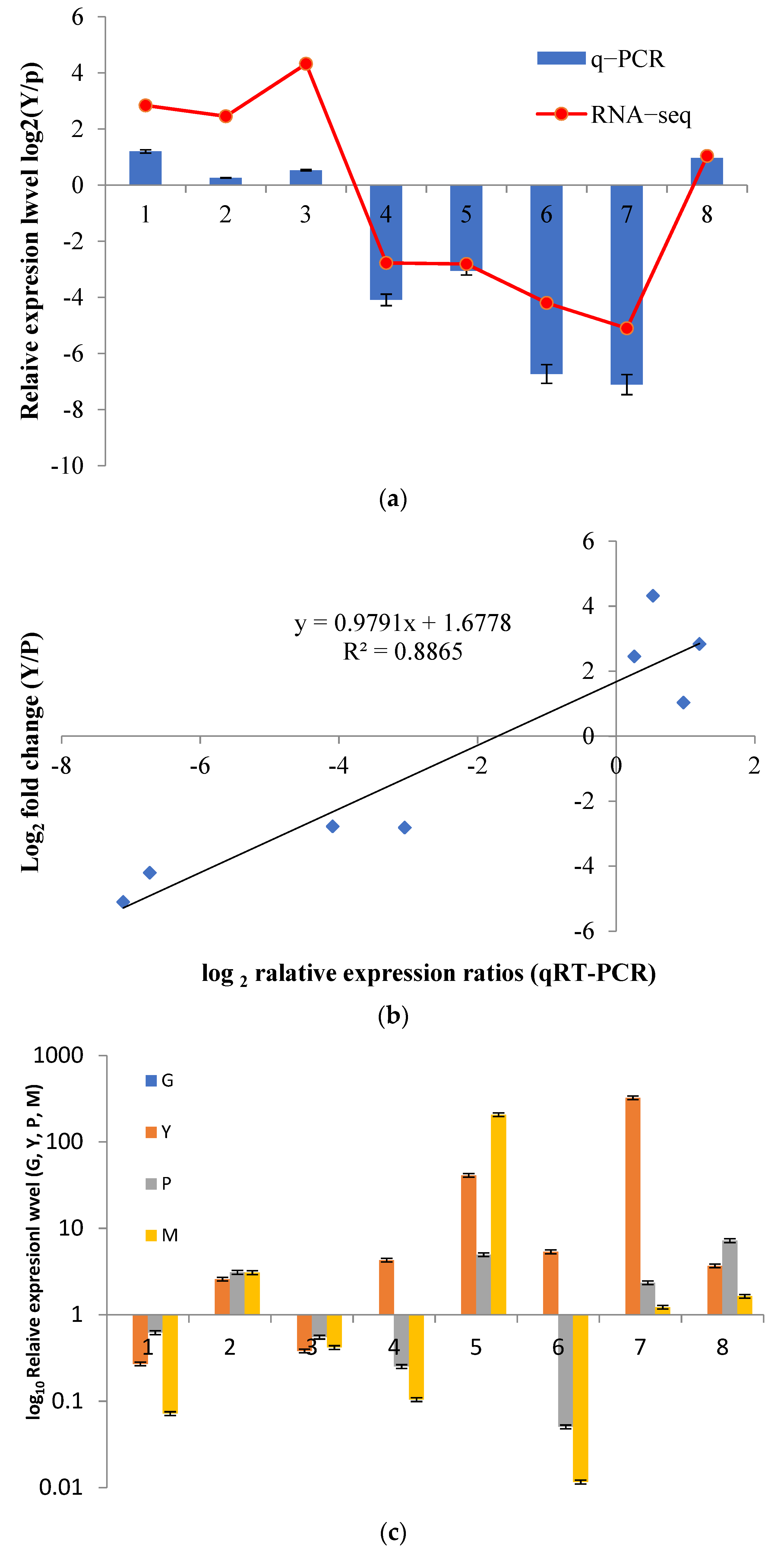
3.7. qRT-PCR
3.8. Regulatory Network of Flavonoid Biosynthesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Alwyn, H. Bignoniaceae: Part II (Tribe Tecomeae). Flora Neotrop. Monogr. 1992, 25, 1–370. [Google Scholar] [CrossRef]
- Grose, S.O.; Olmstead, R.G. Evolution of a Charismatic Neotropical Clade: Molecular Phytogeny of Tabebuia S.L. Crescentieae, and Allied Genera (Bignoniaceae). Syst. Bot. 2007, 32, 650–659. [Google Scholar] [CrossRef]
- Grose, S.O.; Olmstead, R.G. Taxonomic Revisions in the Polyphyletic Genus Tabebuia S. I. (Bignoniaceae). Syst. Bot. 2009, 32, 660–670. [Google Scholar] [CrossRef]
- Kadereit, J. The Families and Genera of Vascular Plants: Volume VII Flowering Plants, Dicotyledons: Lamiales. (Except Acanthaceae Including Avicenniaceae); Springer: Berlin, Germany, 2004; pp. 9–38. [Google Scholar]
- George, W.; Derra, l.R. A Tropical Garden Flora; Bishop Museum Press: Honolulu, HI, USA, 2005. [Google Scholar]
- Wang, X. Botanical Garden Tree Species of Tabebuia Chrysantha. Guangdong For. Sci. Technol. 2007, 23, 115–116. [Google Scholar] [CrossRef]
- Yuan, L.; Wang, S.; Liu, N.; Zhang, Q.; Ren, H.; Lei, Z.; Pan, G.; Luo, J. The Ecological and Biological Characteristics and Sowing Propagation Techniques of Tabebuia chrysantha. Ecol. Sci. 2014, 33, 984–990. [Google Scholar]
- Cai, D.; Fang, L.; Shang, X.; Zhang, P. Research Status and Development Prospects of Tabebuia. Eucalypt Sci. Technol. 2016, 33, 46–50. [Google Scholar]
- Ana, C.D.S.B.; Maria, L.G.; Joelma, F.D.O.F.; Nasser, R.A.; Eric, H.F.F.F.; Mario, B.F. Effects of a Tabebuia avellanedae Extract and Lapachol on the Labeling of Blood Constituents with Technetium-99M. Sci. Res. Essays 2015, 10, 97–104. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.M.; Myung, C.H.; Lee, K.R.; Hyun, S.M.; Lee, J.E.; Park, Y.S.; Jeon, S.R.; Park, J.I.; Chang, S.E.; et al. Melanogenesis Inhibition of β-Lapachone, a Natural Product from Tabebuia avellanedae, with Effective In Vivo Lightening Potency. Arch. Dermatol. Res. 2015, 307, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.P.; Senna Bonfa, I.M.; Cavalcante, R.B.; Okoba, D.; de Souza Vasconcelos, S.B.; Candeloro, L.; de Oliveira Filiu, W.F.; Duenhas Monreal, A.C.; da Silva, V.J.; Santa Rita, P.H.; et al. Tabebuia Aurea Decreases Inflammatory, Myotoxic and Hemorrhagic Activities Induced by the Venom of Bothrops Neuwiedi. J. Ethnopharmacol. 2014, 158, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Koyama, J.; Morita, I.; Tagahara, K.; Hirai, K.J. Cyclopentene Dialdehydes from Tabebuia impetiginosa. Phytochemistry 2000, 53, 869–872. [Google Scholar] [CrossRef]
- De Miranda, F.G.; Vilar, J.C.; Alves, I.A.; Cavalcanti, S.C.; Antoniolli, A.R. Antinociceptive and Antiedematogenic Properties and Acute Toxicity of Tabebuia Avellanedae Lor. Ex Griseb. Inner Bark Aqueous Extract. BMC Pharmacol. 2001, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.E.; Moretti, M.; Budni, J.; Balen, G.O.; Fernandes, S.C.; Veronezi, P.O.; Heller, M.; Micke, G.A.; Pizzolatti, M.G.; Rodrigues, A.L. NMDA Receptors and the l-Arginine-Nitric Oxide-Cyclic Guanosine Monophosphate Pathway Are Implicated in the Antidepressant-Like Action of the Ethanolic Extract from Tabebuia Avellanedaein Mice. J. Med. Food 2013, 16, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Cock, I.E. Tabebuia impetiginosa (Mart. Ex DC. Mattos) Bark Extracts Inhibit the Growth Gastrointestinal Bacterial Pathogens and Potentiate the Activity of Some Conventional Antibiotics. Pharmacogn. Commun. 2020, 10, 75–82. [Google Scholar] [CrossRef]
- Pires, T.C.; Dias, M.I.; Calhelha, R.C.; Carvalho, A.M.; Queiroz, M.J.; Barros, L.; Ferreira, I.C. Bioactive Properties of Tabebuia impetiginosa-Based Phytopreparations and Phytoformulations: A Comparison Between Extracts and Dietary Supplements. Molecules 2015, 20, 22863–22871. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Fukuda, Y.; Tokikura, C.; Noda, M.; Yamamoto, A.; Yamamoto, M.; Yamashita, M.; Zaima, N.; Iida, A.; Moriyama, T. The Anti-obesity Effect of Taheebo (Tabebuia avellanedae Lorentz Ex Griseb) Extract in Ovariectomized Mice and the Identification of a Potential Anti-obesity Compound. Biochem. Biophys. Res. Commun. 2016, 478, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, E.V.; Carneiro, J.W.; Ferreira, V.F. Quantitative Structure-Activity Relationship in Aziridinyl-1,4-Naphthoquinone Antimalarials: Study of Theoretical Correlations by the PM3 Method. Bioorg. Med. Chem. 2004, 12, 87–93. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Plants as a Source of Anti-cancer Agents. J. Ethnopharmacol. 2005, 100, 72–79. [Google Scholar] [CrossRef]
- Byeon, S.E.; Chung, J.Y.; Lee, Y.G.; Kim, B.H.; Kim, K.H.; Cho, J.Y. In Vitro and In Vivo Anti-Inflammatory Effects of Taheebo, a Water Extract from the Inner Bark of Tabebuia avellanedae. J. Ethnopharmacol. 2008, 119, 145–152. [Google Scholar] [CrossRef]
- Twardowschy, A.; Freitas, C.S.; Baggio, C.H.; Mayer, B.; dos Santos, A.C.; Pizzolatti, M.G.; Zacarias, A.A.; dos Santos, E.P.; Otuki, M.F.; Marques, M.C. Antiulcerogenic Activity of Bark Extract of Tabebuia Avellanedae, Lorentz Ex Griseb. J. Ethnopharmacol. 2008, 118, 455–459. [Google Scholar] [CrossRef]
- Malange, K.F.; Dos Santos, G.G.; Kato, N.N.; Toffoli-Kadri, M.C.; Carollo, C.A.; Silva, D.B.; Portugal, L.C.; Alves, F.M.; Rita, P.H.S.; Parada, C.A.; et al. Tabebuia Aurea Decreases Hyperalgesia and Neuronal Injury Induced by Snake Venom. J. Ethnopharmacol. 2019, 233, 131–140. [Google Scholar] [CrossRef]
- Mistrangelo, M.; Cornaglia, S.; Pizzio, M.; Rimonda, R.; Gavello, G.; Dal Conte, I.; Mussa, A. Immunostimulation to Reduce Recurrence after Surgery for Anal Condyloma Acuminata: A Prospective Randomized Controlled Trial. Colorectal Dis. 2010, 12, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Jeong, K.M.; Lee, J.; Zhao, J.; Choi, S.Y.; Baek, K.S. Development and Validation of an Analytical Method Readily Applicable for Quality Control of Tabebuia impetiginosa (Taheebo) Ethanolic Extract. J. AOAC Int. 2018, 101, 695–700. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Rai, A.; Saito, K.; Yamazaki, M. Integrated Omics Analysis of Specialized Metabolism in Medicinal Plants. Plant J. 2017, 90, 764–787. [Google Scholar] [CrossRef]
- Riaño-Pachón, D.M.; Nagel, A.; Neigenfind, J.; Wagner, R.; Basekow, R.; Weber, E.; Mueller-Roeber, B.; Diehl, S.; Kersten, B. GabiPD: The GABI Primary Database-A Plant Integrative ‘Omics’ Database. Nucleic Acids Res. 2009, 37, D954–D959. [Google Scholar] [CrossRef]
- Shen, Q.; Fu, L.; Dai, F.; Jiang, L.; Zhang, G.; Wu, D. Multi-omics Analysis Reveals Molecular Mechanisms of Shoot Adaption to Salt Stress in Tibetan Wild Barley. BMC Genom. 2016, 17, 889. [Google Scholar] [CrossRef]
- Villar, M.; Ayllón, N.; Alberdi, P.; Moreno, A.; Moreno, M.; Tobes, R.; Mateos-Hernández, L.; Weisheit, S.; Bell-Sakyi, L.; de la Fuente, J. Integrated Metabolomics, Transcriptomics and Proteomics Identifies Metabolic Pathways Affected by Anaplasma phagocytophilum Infection in Tick Cells. Mol. Cell. Proteom. 2015, 14, 3154–3172. [Google Scholar] [CrossRef]
- Vasilev, N.; Boccard, J.; Lang, G.; Grömping, U.; Fischer, R.; Goepfert, S.; Rudaz, S.; Schillberg, S. Structured Plant Metabolomics for the Simultaneous Exploration of Multiple Factors. Sci. Rep. 2016, 6, 37390. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Weng, K.; Guo, Y.; Peng, J.; Zhu, Z. An Integrated Targeted Metabolomic Platform for High-Throughput Metabolite Profiling and Automated Data Processing. Metabolomics 2015, 11, 1575–1586. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for Large-Scale Metabolic Profiling of Serum and Plasma Using Gas Chromatography and Liquid Chromatography Coupled to Mass Spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global Metabolic Profiling Procedures for Urine Using UPLC-MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef]
- Zelena, E.; Dunn, W.B.; Broadhurst, D.; Francis-McIntyre, S.; Carroll, K.M.; Begley, P.; O’Hagan, S.; Knowles, J.D.; Halsall, A.; HUSERMET Consortium; et al. Development of a Robust and Repeatable UPLC-MS Method for the Long-Term Metabolomic Study of Human Serum. Anal. Chem. 2009, 81, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global Metabolic Profiling of Animal and Human Tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, T.; Shen, X.; Liu, J.; Zhao, D.; Sun, Y.; Wang, L.; Liu, Y.; Gong, X.; Liu, Y.; et al. Serum Metabolomics for Early Diagnosis of Esophageal Squamous Cell Carcinoma by UHPLC-QTOF/MS. Metabolomics 2016, 12, 116. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Feature Counts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript Assembly and Quantification by RNA-Seq Reveals Unannotated Transcripts and Isoform Switching During Cell Differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated Genome Annotation and Pathway Identification Using the KEGG Orthology (KO) as a Controlled Vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Hauke, J.; Kossowski, T. Comparison of Values of Pearson’s and Spearman’s Correlation Coefficients on the Same Sets of Data. Quaest. Geogr. 2011, 30, 87–93. [Google Scholar] [CrossRef]
- Kumari, S.; Nie, J.; Chen, H.S.; Ma, H.; Stewart, R.; Li, X.; Lu, M.Z.; Taylor, W.M.; Wei, H. Evaluation of Gene Association Methods for coexpression Network Construction and Biological Knowledge Discovery. PLoS ONE 2012, 7, e50411. [Google Scholar] [CrossRef]
- Rebekić, A.; Lončarić, Z.; Petrović, S.; Marić, S. Pearson’s or Spearman’s Correlation Coefficient-Which One to Use? Poljoprivreda 2015, 21, 47–54. [Google Scholar] [CrossRef]
- S Song, T.; Xu, H.; Sun, N.; Jiang, L.; Tian, P.; Yong, Y.; Yang, W.; Cai, H.; Cui, G. Metabolomic Analysis of Alfalfa (Medicago Sativa L.) Root-Symbiotic Rhizobia Responses under Alkali Stress. Front. Plant Sci. 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for Linking Genomes to Life and the Environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.M.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and Biochemistry of Seed Flavonoids. Annu. Rev. Plant Biol. 2006, 57, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Qiao, Z.; Wang, H.; Zhu, L.; Zhang, L. Flavonoids: Promising Anticancer Agents. Med. Res. Rev. 2003, 23, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Cushnie, T.P.T.; Lamb, A.J. Antimicrobial Activity of Flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef]
- Ji, X.; Ren, J.; Lang, S.; Wang, D.; Zhu, L.; Song, X. Differential Regulation of Anthocyanins in Cerasus humilis Fruit Color Revealed by Combined Transcriptome and Metabolome Analysis. Forests 2020, 11, 1065. [Google Scholar] [CrossRef]
- Shen, Z.; Xu, Y.; Xia, B.; Li, J. Transcription Factors Involved in Plant Flavonoid Biosynthesis of Secondary Metabolism and Its Application in Genetic Engineering. Mol. Plant Breed. 2008, 16, 542–548. [Google Scholar] [CrossRef]
- Sharma, P.K.; Khanna, R.N.; Rohatgi, B.K.; Thomson, R.H. Tecomaquinone-III: A New Quinone from Tabebuia pentaphylla. Phytochemistry 1988, 27, 632–633. [Google Scholar] [CrossRef]
- Manners, G.D.; Jurd, L. A New Naphthaquinone from Tabebuia guayacan. Phytochemistry 1976, 15, 225–226. [Google Scholar] [CrossRef]
- Blatt, C.T.T.; Salatino, A.; Salatino, M.L.F. Flavonoids of Tabebuia caraiba (Bignoniaceae). Biochem. Syst. Ecol. 1996, 24, 89. [Google Scholar] [CrossRef]
- Wagner, H.; Kreher, B.; Lotter, H.; Hamburger, M.O.; Cordell, G.A. Structure Determination of New Isomeric Naphtho [2,3-b] Furan-4,9-Diones from Tabebuia Avellanedae by the Selective-INEPT Technique. Helv. Chim. Acta 1989, 72, 659–667. [Google Scholar] [CrossRef]
- Warashina, T.; Nagatani, Y.; Noro, T. Constituents from the Bark of Tabebuia impetiginosa. Phytochemistry 2004, 65, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Lee, K.G.; Shibamoto, T.; Lee, S.E.; Takeoka, G.R. Antioxidant Activity and Characterization of v Olatile Constituents of Taheebo (Tabebuia impetiginosa Martius Ex DC). J. Agric. Food Chem. 2003, 51, 295–300. [Google Scholar] [CrossRef]
- Zhang, L.; Hasegawa, I.; Ohta, T. Anti-Inflammatory Cyclopentene Derivatives from the Inner Bark of Tabebuia Avellanedae. Fitoterapia 2016, 109, 217–223. [Google Scholar] [CrossRef]
- Yonekura-Sakakibara, K.; Higashi, Y.; Nakabayashi, R. The Origin and Evolution of Plant Flavonoid Metabolism. Front. Plant Sci. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, A.; Minihane, A.M. The Role of Metabolism (and the Microbiome) in Defining the Clinical Efficacy of Dietary Flavonoids. Am. J. Clin. Nutr. 2017, 105, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Takahashi, S.; Waki, T. Formation of Flavonoid Metabolons: Functional Significance of Protein-Protein Interactions and Impact on Flavonoid Chemodiversity. Front. Plant Sci. 2019, 10, 821. [Google Scholar] [CrossRef]
- Farzaei, M.H.; Singh, A.K.; Kumar, R.; Croley, C.R.; Pandey, A.K.; Coy-Barrera, E.; Kumar Patra, J.; Das, G.; Kerry, R.G.; Annunziata, G.; et al. Targeting Inflammation by Flavonoids: Novel Therapeutic Strategy for Metabolic Disorders. Int. J. Mol. Sci. 2019, 20, 4957. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Spagnuolo, C.; Tedesco, I.; Bilotto, S.; Russo, G.L. The Flavonoid Quercetin in Disease Prevention and Therapy: Facts and Fancies. Biochem. Pharmacol. 2012, 83, 6–15. [Google Scholar] [CrossRef]
- Bischoff, S.C. Quercetin: Potentials in the Prevention and Therapy of Disease. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, K.; Dwyer-Nield, L.D.; Serkova, N.J.; Hasebroock, K.M.; Tyagi, A.; Raina, K.; Singh, R.P.; Malkinson, A.M.; Agarwal, R. Silibinin Prevents Lung Tumorigenesis in Wild-Type but Not in iNOS-/-Mice: Potential of Real-Time Micro-CT in Lung Cancer Chemoprevention Studies. Clin. Cancer Res. 2011, 17, 753–761. [Google Scholar] [CrossRef]
- Tyagi, A.; Agarwal, C.; Harrison, G.; Glode, L.M.; Agarwal, R. Silibinin Causes Cell Cycle Arrest and Apoptosis in Human Bladder Transitional Cell Carcinoma Cells by Regulating CDKI-CDK-Cyclin Cascade, and Caspase 3 and PARP Cleavages. Carcinogenesis 2004, 25, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Su, J. Transcriptomic and Metabolomics Analysis Reveals the Biosynthesis and Distribution of Active Ingredients in Nanhaia speciosa; Guangzhou University of Chinese Medicine: Guangzhou, China, 2021; pp. 5–7. [Google Scholar]
- Spitz, F.; Furlong, E.E. Transcription Factors: From Enhancer Binding to Developmental Control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Mathews, H.; Clendennen, S.K.; Caldwell, C.G.; Liu, X.L.; Connors, K.; Matheis, N.; Schuster, D.K.; Menasco, D.J.; Wagoner, W.; Lightner, J.; et al. Activation Tagging in Tomato Identifies a Transcriptional Regulator of Anthocyanin Biosynthesis, Modification, and Transport. Plant Cell 2003, 15, 1689–1703. [Google Scholar] [CrossRef]
- Goodrich, J.; Carpenter, R.; Coen, E.S. A Common Gene Regulates Pigmentation Pattern in Diverse Plant Species. Cell 1992, 68, 955–964. [Google Scholar] [CrossRef]
- Sagasser, M.; Lu, G.H.; Hahlbrock, K.; Weisshaar, B.A.A. Tshaliana TRANSPARENT TESTA 1 Is Involved in Seed Coat Development and Defines the WIP Subfamily of Plant Zinc Finger Proteins. Genes Dev. 2002, 16, 138–149. [Google Scholar] [CrossRef]
- Selinger, D.A.; Chandler, V.L. A Mutation in the Pale Aleurone color1 Gene Identifies a Novel Regulator of the Maize Anthocyanin Pathway. Plant Cell 1999, 11, 5–14. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Formula | Class | 1VIP | Fold-Change (FC) | log2FC |
|---|---|---|---|---|---|
| Gibberellin A53 | C20H28O5 | Isoprenoids | 1.12 | 568 | −9.15 |
| Silibinin | C25H22O10 | Flavonoids | 1.08 | 307 | −8.26 |
| Cucurbitacin B | C32H46O8 | Sterols | 1.34 | 164 | −7.35 |
| 26-Hydroxyecdysone | C27H44O7 | Sterols | 1.24 | 155 | −7.28 |
| Warfarin | C19H16O4 | Coumarins and derivatives | 1.34 | 131 | −7.03 |
| Chalconaringenin | C15H12O5 | Flavonoids | 1.28 | 112 | −6.81 |
| N-Acetylaspartylglutamate | C11H16N2O8 | Sterols | 1.18 | 99 | −6.63 |
| Benzoate | C7H6O2 | Benzenoids | 1.05 | 86 | −6.42 |
| Phytosphingosine | C18H39NO3 | Organonitrogen compounds | 1.06 | 71 | −6.14 |
| Epicatechin | C6H6N2O | Flavonoids | 1.10 | 67 | −6.06 |
| D-Xylonate | C5H10O6 | Organooxygen compounds | 1.18 | 180 | 7.49 |
| Geranylgeraniol | C20H34O | Isoprenoids | 1.29 | 182 | 7.51 |
| Indoleacetic acid | C10H9NO2 | Indoles and derivatives | 1.32 | 211 | 7.72 |
| Cyanidin 3-glucoside | C21H21O11 | Flavonoids | 1.00 | 213 | 7.74 |
| Prunasin | C14H17NO6 | Benzene and substituted derivatives | 1.38 | 225 | 7.82 |
| Geniposidic acid | C16H22O10 | Isoprenoids | 1.01 | 278 | 8.12 |
| Astringin | C20H22O9 | Aromatic polyketides | 1.25 | 482 | 8.91 |
| Peonidin−3-glucoside | C22H23O11 | Flavonoids | 1.17 | 514 | 9.00 |
| Malvidin 3-glucoside | C23H25O12 | Flavonoids | 1.34 | 1554 | 10.60 |
| Mycophenolic acid O-acyl-glucuronide | C23H28O12 | Organooxygen compounds | 1.11 | 3239 | 11.66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, X.; Liu, G.; Wu, Z.; Zhang, P. Combined Transcriptomic and Metabolomic Analysis Reveals the Mechanism of Flavonoid Biosynthesis in Handroanthus chrysanthus (Jacq.) S.O.Grose. Forests 2022, 13, 1285. https://doi.org/10.3390/f13081285
Shang X, Liu G, Wu Z, Zhang P. Combined Transcriptomic and Metabolomic Analysis Reveals the Mechanism of Flavonoid Biosynthesis in Handroanthus chrysanthus (Jacq.) S.O.Grose. Forests. 2022; 13(8):1285. https://doi.org/10.3390/f13081285
Chicago/Turabian StyleShang, Xiuhua, Guo Liu, Zhihua Wu, and Peijian Zhang. 2022. "Combined Transcriptomic and Metabolomic Analysis Reveals the Mechanism of Flavonoid Biosynthesis in Handroanthus chrysanthus (Jacq.) S.O.Grose" Forests 13, no. 8: 1285. https://doi.org/10.3390/f13081285