
Full-Length Transcriptome Sequencing and Identification of Genes Related to Terpenoid Biosynthesis in Cinnamomum migao H. W. Li
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Construction, and Transcriptome Sequencing
2.3. Raw Data Analysis
2.4. SSR, CDS, TFs, and LncRNA Prediction
2.5. Functional Annotation of Transcripts
2.6. DETs Analysis
2.7. RT-qPCR Analyses
3. Results
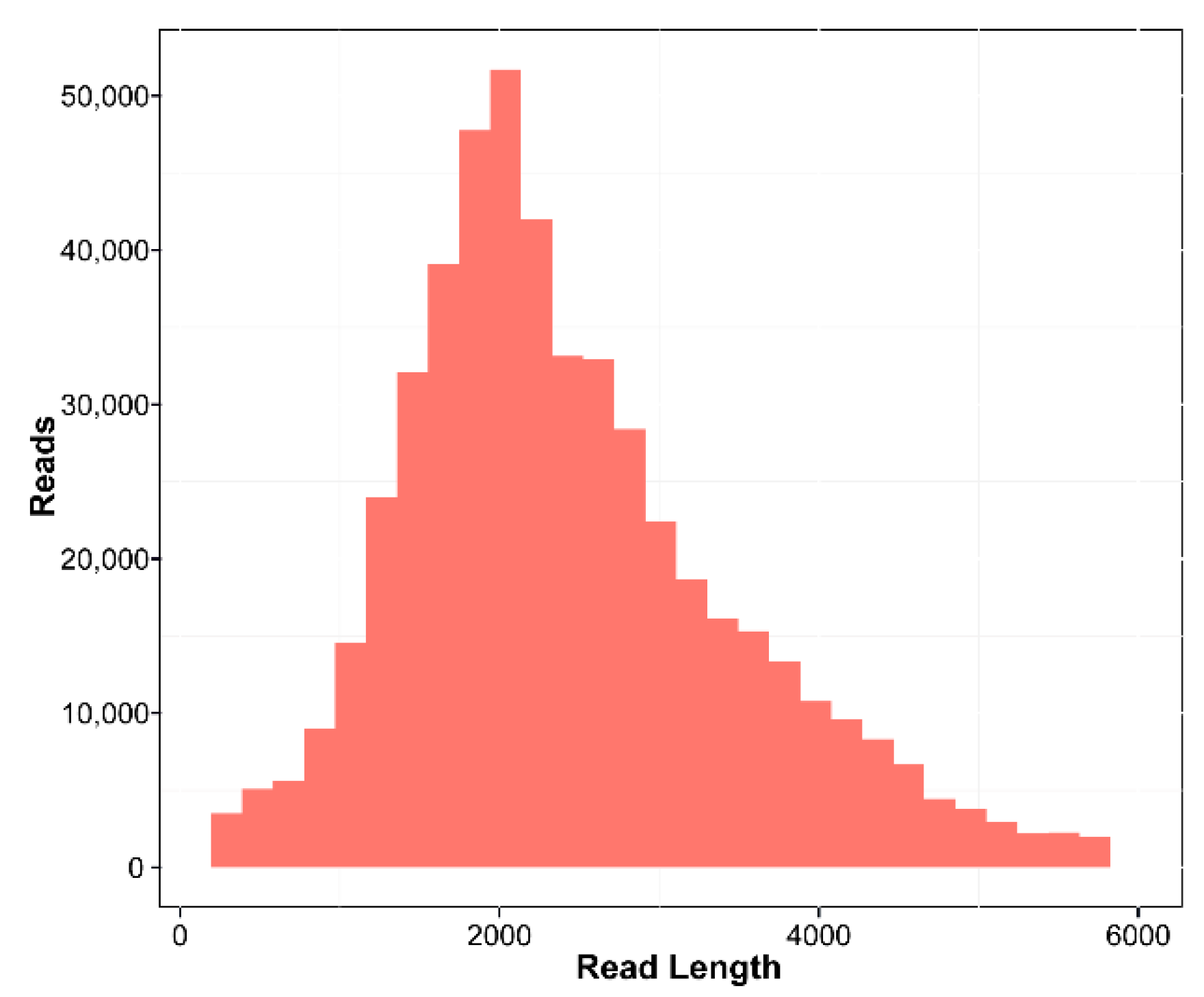
3.1. Transcriptome Analysis
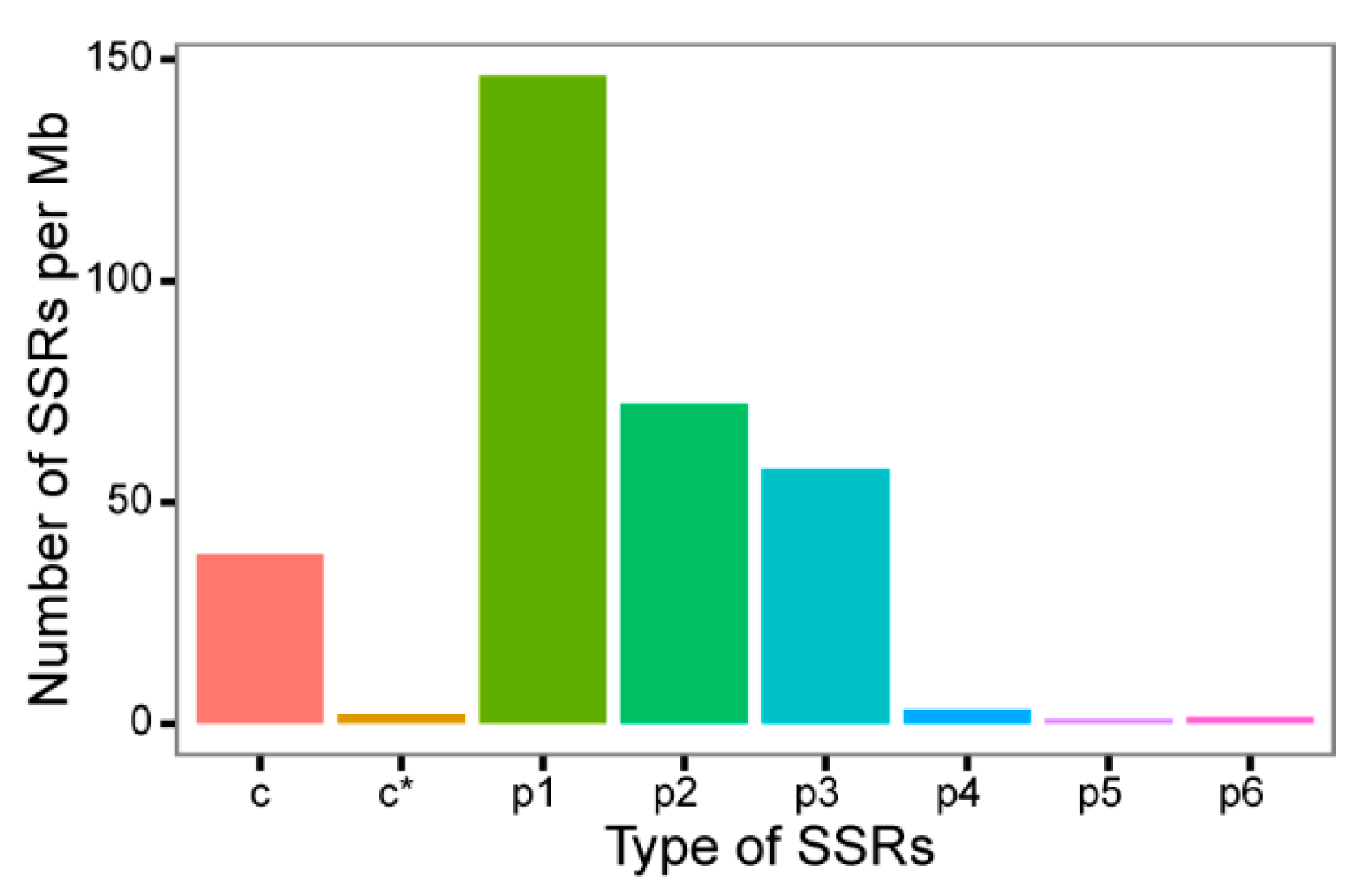
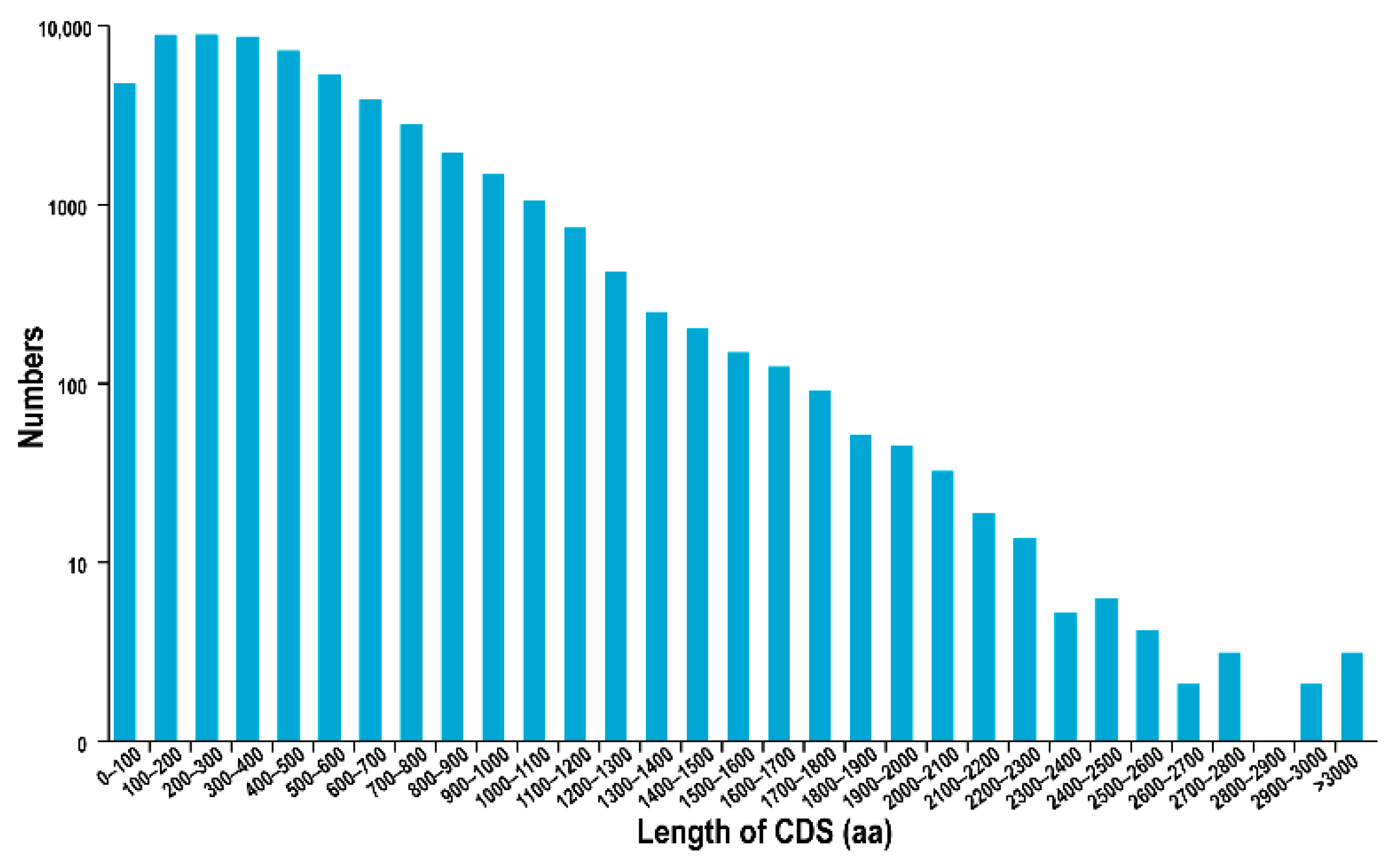
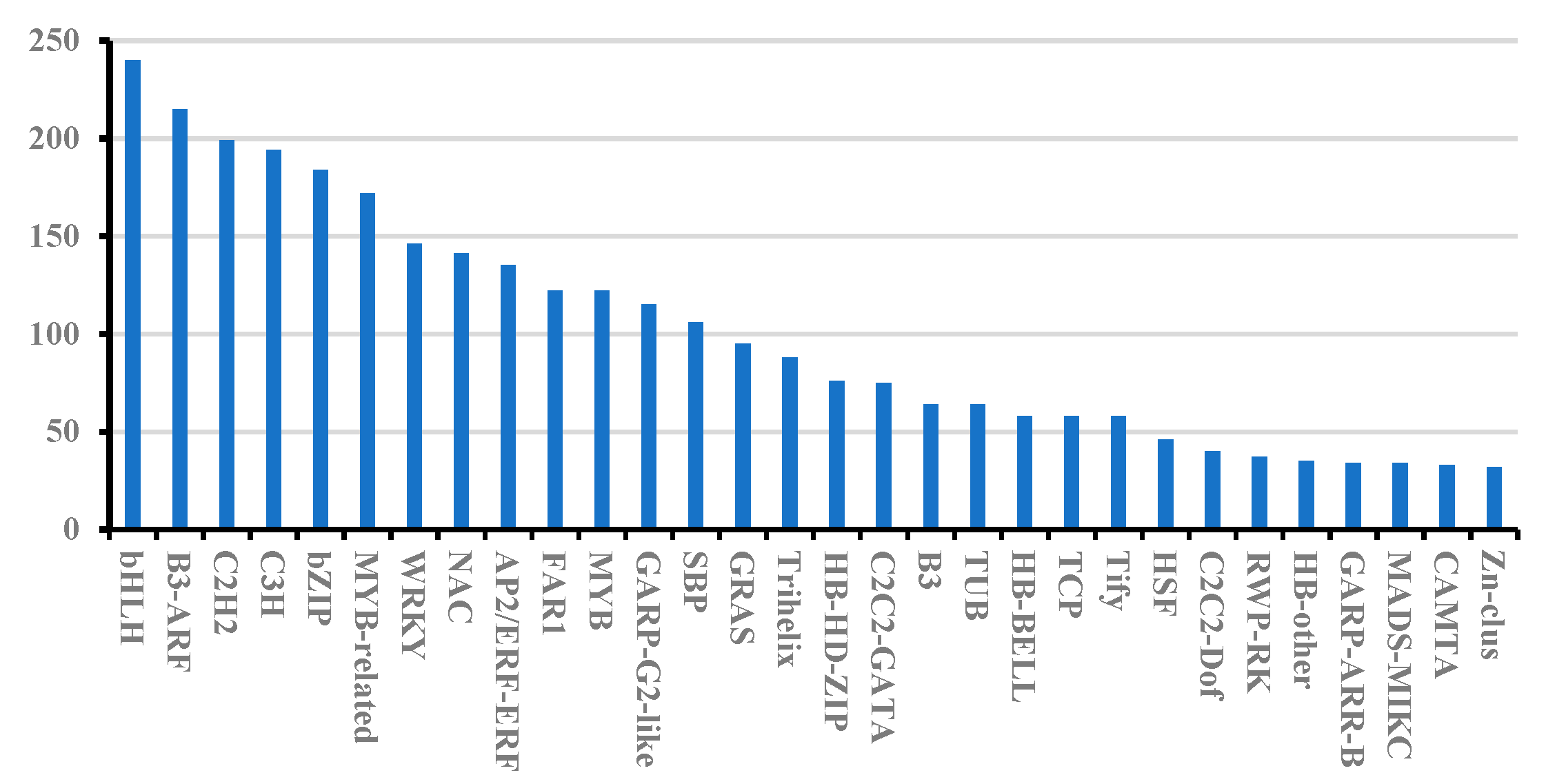
3.2. SSR, CDS, TFs, and LncRNA Prediction
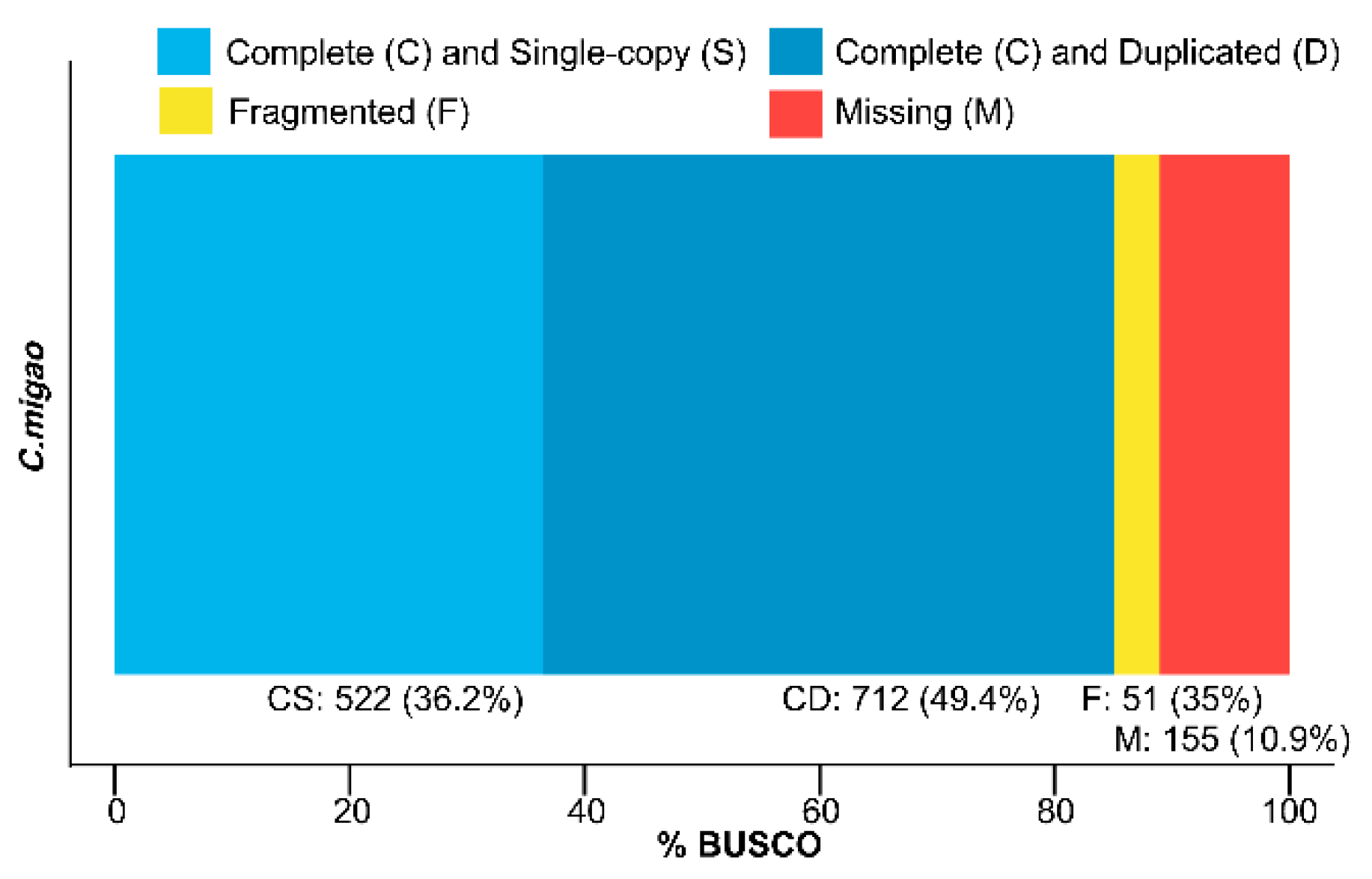
3.3. Functional Annotation
3.4. Identification of DETs in C. migao Fruits at Different Developmental Stages
3.5. Analysis of DETs Involved in Terpenoid Biosynthesis
3.6. RT-qPCR of DETs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shi, H.Y.; Shi, Q.L.; Wang, T.W.; Qin, J.Z.; Wang, Y.P. Migao-a special Miao medicinal plants in Guizhou Province. In Proceedings of the National Symposium on Miao Medicine, Guiyang, China, 1 November 2003. [Google Scholar]
- Zheng, Y.Y.; Qiu, D.W.; Liang, G.Y.; Sun, X.H.; Sui, Y.H. Research and Industrialization of “Da Guo Mu Jiang Zi” in Guizhou Province. World Sci. Technol. 2005, 02, 112–114. [Google Scholar]
- Wang, J.C.; Liu, J.M.; Wen, A.H.; Gao, P. Research progress on the medicinal plant “Da Guo Mu Jiang Zi” in Guizhou Province. Heilongjiang Agric. Sci. 2015, 5, 157–160. [Google Scholar]
- Guo, J.T.; Zhang, Y.P.; Liu, J.; Xu, J.; Cheng, C.; Liu, Y. Comparison of volatile oil composition and antioxidant activity in different parts of Lisea lancilimba Merr. Sci. Technol. Food Ind. 2023, 44, 306–315. [Google Scholar]
- Liu, J.; Guo, J.T.; Liu, Y.; Cheng, C.; Huang, K.; Jian, L.N.; Xu, J.; Zhang, Y.P. Extraction optimization, composition analysis of volatile oil from Lisea lancilimba Merr. and its antioxidant activity. Sci. Technol. Food Ind. 2022, 9, 211–219. [Google Scholar]
- Sun, H.C.; Li, J.; Zhou, Z.H.; Wang, Z.D.; Dong, H.; Tang, X.Q.; Liu, W.Q.; Zhang, L.Y. Research progress in chemical constituents, pharmacological effect and industrialization of Lisea lancilimba Merr. Cent. South Pharm. 2022, 20, 668–671. [Google Scholar]
- Yan, T.; Zhou, Z.Y.; Luo, D.Y.; Chi, M.Y.; Wang, A.M.; Huang, Y.; Zheng, L. GC-MS analysis of three different methods for extracting volatile oil components from fresh and dry fruits of Cinnamomum migao. J. Chin. Med. Mater. 2022, 1, 123–129. [Google Scholar]
- Chen, S.Y.; Tang, Z.X.; Yang, X.; Tu, X.H.; Yuan, L.F.; Pan, S.J.; Deng, W.J.; Zhao, T.T. Study on the molecular mechanism of main volatile components of Lisea lancilimba Merr. Chin. J. Ethnomed. Ethnopharmacol. 2022, 31, 33–38. [Google Scholar]
- Huang, K.; Liu, J.; Huang, C.H.; Liu, Y.; Cheng, C.; Zhang, Y.P.; Xu, J. Comparative analysis of volatile oil and fatty oil constituents from Cinnamomum migao in different sources. China Pharm. 2020, 31, 1961–1966. [Google Scholar]
- Luo, J.; Zhu, D.; Liao, X.; Huang, J.; Tang, J.; Liu, W.; Bao, J.P.; Tan, J.H. Comparative research on the components of volatile oils in Listea Lam based on the theory of using fresh materials in Miao medicine. Lishizhen Med. Mater. Res. 2019, 20, 574–576. [Google Scholar]
- Chen, J.Z.; Huang, X.L.; Xiao, X.F.; Liu, J.M.; Liao, X.F.; Sun, Q.W.; Peng, L.; Zhang, L. Seed Dormancy Release and Germination Requirements of Cinnamomum migao, an Endangered and Rare Woody Plant in Southwest China. Front. Plant Sci. 2022, 13, 770940. [Google Scholar] [CrossRef]
- Huang, X.L.; Tian, T.; Chen, J.Z.; Wang, D.; Tong, B.L.; Liu, J.M. Transcriptome analysis of Cinnamomum migao seed germination in medicinal plants of southwest China. BMC Plant Biol. 2021, 21, 270. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Su, W.; Hussain, M.A.; Mehmood, S.S.; Zhang, X.; Cheng, Y.; Zou, X.; Lv, Y. Integrated Analysis of Metabolome and Transcriptome Reveals Insights for Cold Tolerance in Rapeseed (Brassica napus L.). Front. Plant Sci. 2021, 12, 721681. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Wang, L.G.; Park, H.J.; Dasari, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression mode. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein family database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated NR Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Qi, H.; Luan, X.; Xu, W.; Yu, F.; Zhong, Y.; Xu, M. The chromosome-level genome sequence of the camphor tree provides insights into Lauraceae evolution and terpene biosynthesis. Plant Biotech. J. 2022, 20, 244–246. [Google Scholar] [CrossRef]
- Jiang, R.; Chen, X.; Liao, X.; Peng, D.; Han, X.; Zhu, C.; Wang, P.; Hufnagel, D.E.; Wang, L.; Li, K.; et al. A Chromosome-Level Genome of the Camphor Tree and the Underlying Genetic and Climatic Factors for Its Top-Geoherbalism. Front. Plant Sci. 2022, 13, 827890. [Google Scholar] [CrossRef]
- Wang, X.D.; Xu, C.Y.; Zheng, Y.J.; Wu, Y.F.; Zhang, Y.T.; Zhang, T.; Xiong, Z.Y.; Yang, H.K.; Li, J.; Fu, C.; et al. Chromosome-level genome assembly and resequencing of camphor tree (Cinnamomum camphora) provides insight into phylogeny and diversification of terpenoid and triglyceride biosynthesis of Cinnamomum. Hortic. Res. 2022, 9, uhac216. [Google Scholar] [CrossRef]
- Li, D.; Lin, H.Y.; Wang, X.; Bi, B.; Gao, Y.; Shao, L.; Zhang, R.; Liang, Y.; Xia, Y.; Zhao, Y.P.; et al. Genome and whole-genome resequencing of Cinnamomum camphora elucidate its dominance in subtropical urban landscapes. BMC Biol. 2023, 21, 192. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Wang, Y.H.; Shen, S.K. Transcriptome Analysis of Cinnamomum chago: A Revelation of Candidate Genes for Abiotic Stress Response and Terpenoid and Fatty Acid Biosyntheses. Front. Genet. 2018, 9, 505. [Google Scholar] [CrossRef]
- Zhao, X.; Yan, Y.; Zhou, W.H.; Feng, R.Z.; Shuai, Y.K.; Yang, L.; Liu, M.J.; He, X.Y.; Wei, Q. Transcriptome and metabolome reveal the accumulation of secondary metabolites in different varieties of Cinnamomum longepaniculatum. BMC Plant Biol. 2022, 22, 243. [Google Scholar] [CrossRef]
- Gao, H.; Zhang, H.; Hu, Y.; Xu, D.; Zheng, S.; Su, S.; Yang, Q. De Novo transcriptome assembly and metabolomic analysis of three tissue types in Cinnamomum cassia. Chin. Herb. Med. 2023, 15, 310–316. [Google Scholar] [CrossRef]
- Guo, S.; Liang, J.; Deng, Z.; Lu, Z.; Fu, M.; Su, J. Full-Length Transcriptome Sequencing Combined with RNA-Seq to Analyze Genes Related to Terpenoid Biosynthesis in Cinnamomum burmannii. Curr. Issues Mol. Biol. 2022, 44, 4197–4215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.V.S.K.; Gu, L.; Reddy, A.S.N. Analysis of Transcriptome and Epitranscriptome in Plants Using PacBio Iso-Seq and Nanopore-Based Direct RNA Sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef]
- He, Z.; Su, Y.; Wang, T. Full-Length Transcriptome Analysis of Four Different Tissues of Cephalotaxus oliveri. Int. J. Mol. Sci. 2021, 22, 787. [Google Scholar] [CrossRef]
- Yin, M.; Chu, S.; Shan, T.; Zha, L.; Peng, H. Full-length transcriptome sequences by a combination of sequencing platforms applied to isoflavonoid and triterpenoid saponin biosynthesis of Astragalus mongholicus Bunge. Plant Methods 2021, 17, 61. [Google Scholar] [CrossRef]
- Vranová, E.; Coman, D.; Gruissem, W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Lou, G.; Zhang, L.; Huang, Y.; Yang, J.; Guo, J.; Qi, Z.; Li, Z.; Zhang, G.; Xu, S.; et al. Unveiling the spatial distribution and molecular mechanisms of terpenoid biosynthesis in Salvia miltiorrhiza and S. grandifolia using multi-omics and DESI-MSI. Hortic. Res. 2023, 10, uhad109. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; DeBose-Boyd, R.A. Post-Translational Regulation of HMG CoA Reductase. CSH. Perspect. Biol. 2022, 14, a041253. [Google Scholar] [CrossRef] [PubMed]
- Wanke, M.; Skorupinska-Tudek, K.; Swiezewska, E. Isoprenoid biosynthesis via 1-deoxy-D-xylulose 5-phosphate/2-C-methyl-D-erythritol 4-phosphate (DOXP/MEP) pathway. Acta Biochim. Pol. 2001, 48, 663–672. [Google Scholar] [CrossRef]
- Jia, Q.; Brown, R.; Köllner, T.G.; Fu, J.; Chen, X.; Wong, G.K.; Gershenzon, J.; Peters, R.J.; Chen, F. Origin and early evolution of the plant terpene synthase family. Proc. Natl. Acad. Sci. USA 2022, 119, e2100361119. [Google Scholar] [CrossRef]
- Tholl, D.; Chen, F.; Petri, J.; Gershenzon, J.; Pichersky, E. Two sesquiterpene synthases are responsible for the complex mixture of sesquiterpenes emitted from Arabidopsis flower. Plant J. 2005, 42, 757–771. [Google Scholar] [CrossRef]
- Dornelas, M.C.; Mazzafera, P. A genomic approach to characterization of the Citrus terpene synthase gene family. Genet. Mol. Biol. 2007, 30, 832–840. [Google Scholar] [CrossRef]
- Külheim, C.; Padovan, A.; Hefer, C.; Krause, S.T.; Köllner, T.G.; Myburg, A.A.; Degenhardt, J.; Foley, W.J. The Eucalyptus terpene synthase gene family. BMC Genom. 2015, 16, 450. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.M.; Aubourg, S.; Schouwey, M.B.; Daviet, L.; Schalk, M.; Toub, O.; Lund, S.T.; Bohlmann, J. Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol. 2010, 10, 226. [Google Scholar] [CrossRef]
- Nieuwenhuizen, N.J.; Green, S.A.; Chen, X.; Bailleul, E.J.; Matich, A.J.; Wang, M.Y.; Atkinson, R.G. Functional genomics reveals that a compact terpene synthase gene family can account for terpene volatile production in apple. Plant Physiol. 2013, 161, 787–804. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Read (Gb) | cDNA Size | CCS Number | Read (Bases) | Mean Read Length |
|---|---|---|---|---|---|
| C. migao | 39.9 | 1–6 k | 515,929 | 1,289,205,518 | 2498 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, Z.; Gong, Q.; Liang, L.; Kong, D.; Zhou, T.; Sun, W.; Pang, Y.; Zhang, Y. Full-Length Transcriptome Sequencing and Identification of Genes Related to Terpenoid Biosynthesis in Cinnamomum migao H. W. Li. Forests 2023, 14, 2075. https://doi.org/10.3390/f14102075
Ju Z, Gong Q, Liang L, Kong D, Zhou T, Sun W, Pang Y, Zhang Y. Full-Length Transcriptome Sequencing and Identification of Genes Related to Terpenoid Biosynthesis in Cinnamomum migao H. W. Li. Forests. 2023; 14(10):2075. https://doi.org/10.3390/f14102075
Chicago/Turabian StyleJu, Zhigang, Qiuling Gong, Lin Liang, Dejing Kong, Tao Zhou, Wei Sun, Yuxin Pang, and Yongping Zhang. 2023. "Full-Length Transcriptome Sequencing and Identification of Genes Related to Terpenoid Biosynthesis in Cinnamomum migao H. W. Li" Forests 14, no. 10: 2075. https://doi.org/10.3390/f14102075