
Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Ilex macrocarpa

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, DNA Extraction, and Sequencing
2.2. Assembly and Annotation of Mitogenomes
2.3. Analysis of Repeated Sequences and Codon Usage
2.4. Analyses of Chloroplast to Mitochondrion DNA Transformation and RNA Editing
2.5. Analysis of Phylogeny and Synteny
3. Results
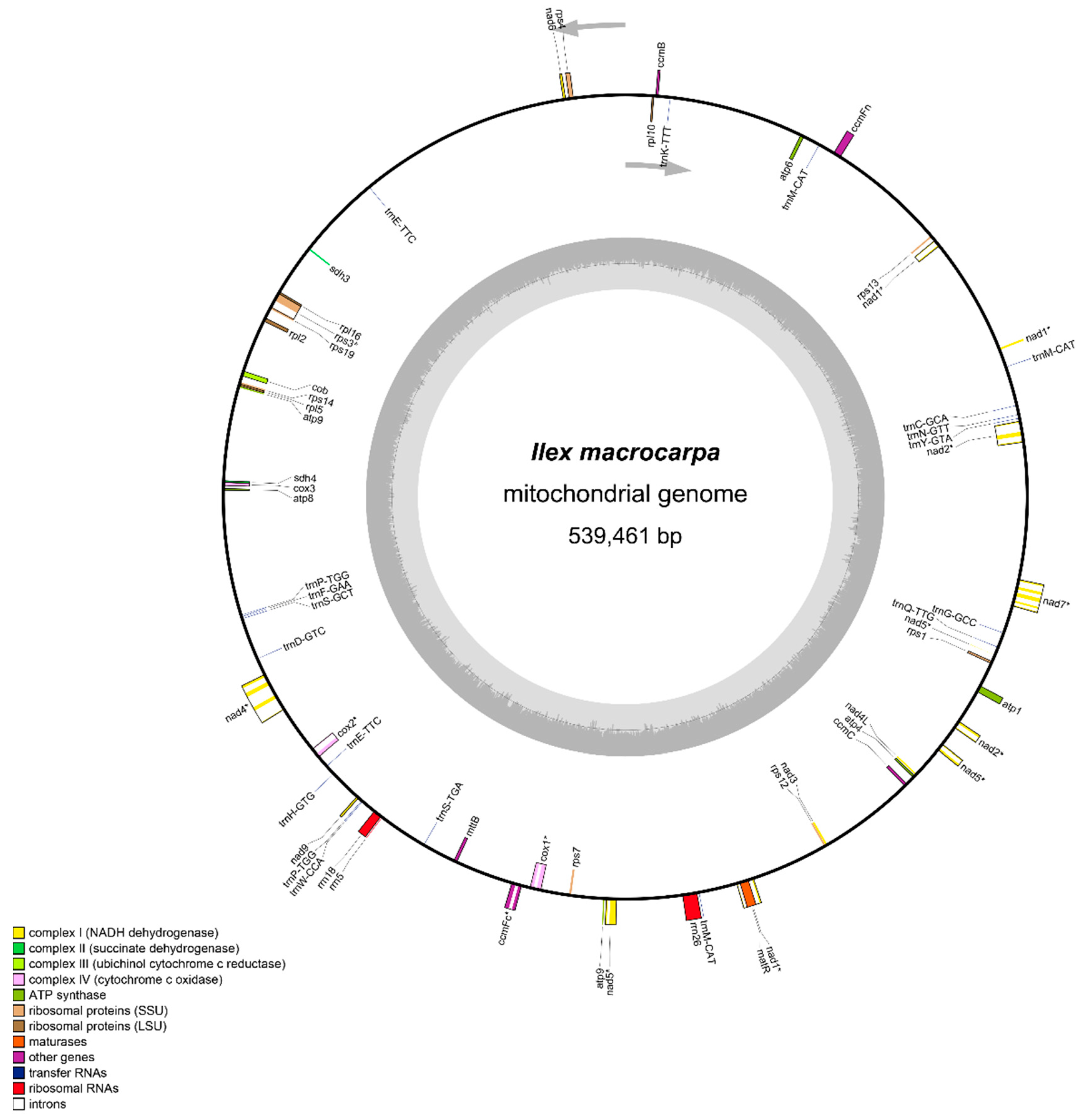
3.1. Features of the I. macrocarpa Mitogenome
3.2. Codon Usage Analysis of PCGs
3.3. Prediction of RNA Editing Sites in PCGs
3.4. Analysis of Repeat Sequences in the I. macrocarpa Mitogenome
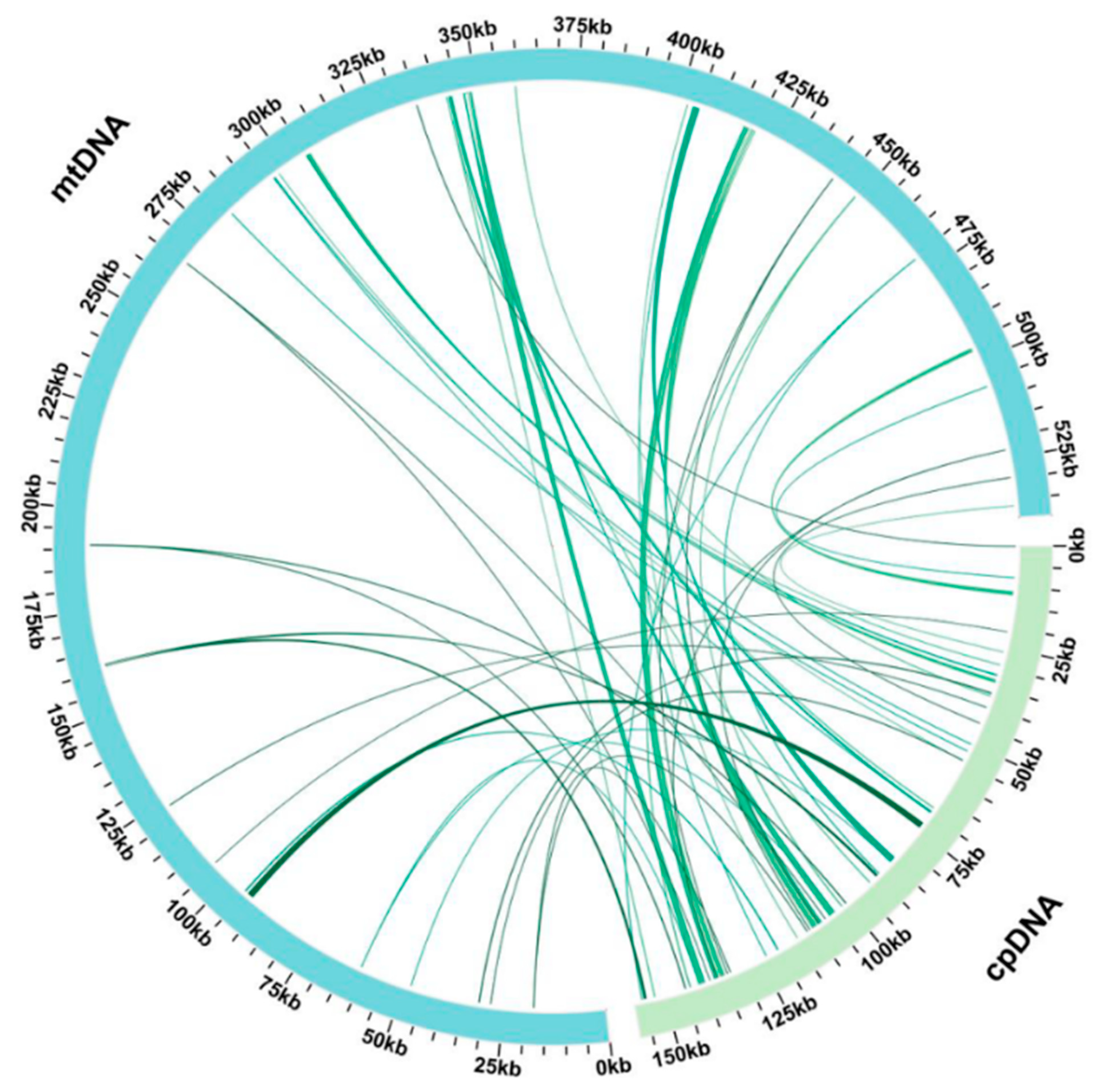
3.5. Chloroplast to Mitochondrion DNA Transfers
3.6. Synteny Sequence Analysis
3.7. Phylogenetic Analysis
4. Discussion
4.1. Characterization of the I. macrocarpa Mitogenome
4.2. Repeated Sequences and RNA Editing
4.3. DNA Fragment Transfer Events
4.4. Analysis of Phylogeny and Synteny
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Z.W.; Zhao, N.; Li, S.S.; Grover, C.E.; Nie, H.S.; Wendel, J.F.; Hua, J. Plant mitochondrial genome evolution and cytoplasmic male sterility. Crit. Rev. Plant Sci. 2017, 36, 55–69. [Google Scholar] [CrossRef]
- Ye, N.; Wang, X.L.; Li, J.; Bi, C.W.; Xu, Y.Q.; Wu, D.Y.; Ye, Q.L. Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. PeerJ 2017, 5, e3148. [Google Scholar] [CrossRef] [PubMed]
- Birky, C. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Nati. Acad. Sci. USA 1995, 92, 11331–11338. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ’master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotf, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Deuk, J.Y.; Park, J.; Kim, J.; Song, W.; Hur, C.G.; Lee, Y.H.; Kang, B.C. Complete sequencing and comparative analyses of the pepper (Capsicum annuum L.) plastome revealed high frequency of tandem repeats and large insertion/deletions on pepper plastome. Plant Cell Rep. 2011, 30, 217–229. [Google Scholar] [CrossRef]
- Petersen, G.; Cuenca, A.; Moller, I.M.; Seberg, O. Massive gene loss in mistletoe (Viscum, Viscaceae) mitochondria. Sci. Rep. 2015, 5, 17588. [Google Scholar] [CrossRef]
- Li, Y.Q.; Zhao, H.K.; Tan, H.; Liu, X.D.; Zhang, C.B.; Dong, Y.S. Analysis and comparison on characteristic of mitochondrial genome of eight plants. Biotechnol. Bull. 2011, 21, 156–162. [Google Scholar] [CrossRef]
- Bi, C.W.; Lu, N.; Xu, Y.Q.; He, C.P.; Lu, Z.H. Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 2020, 21, 3778. [Google Scholar] [CrossRef]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; Mccauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.Y.; Li, S.X.; Bi, C.W.; Hao, Z.D.; Sun, C.R.; Ye, N. Complete chloroplast genome sequence of a major economic species, Ziziphus jujuba (Rhamnaceae). Curr Genet. 2017, 63, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Giege, P.; Brennicke, A. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc. Natl. Acad. Sci. USA 1999, 96, 15324–15329. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Grunheit, N.; Ahmadinejad, N.; Timmis, J.N.; Martin, W. Mutational decay and age of chloroplast and mitochondrial genomes transferred recently to angiosperm nuclear chromosomes. Plant Physiol. 2005, 138, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Turmel, M.; Otis, C.; Lemieux, C. The chloroplast and mitochondrial genome sequences of the charophyte Chaetosphaeridium globosum: Insights into the timing of the events that restructured organelle DNAs within the green algal lineage that led to land plants. Proc. Natl. Acad. Sci. USA 2002, 99, 11275–11280. [Google Scholar] [CrossRef]
- Wang, D.; Wu, Y.W.; Shih, A.C.; Wu, C.S.; Wang, Y.N.; Chaw, S.M. Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 mya. Mol. Biol. Evol. 2007, 24, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Z.; Cao, D.D.; Li, S.S.; Su, A.G.; Geng, J.N.; Grover, C.E.; Hu, S.N.; Hua, J.P. The complete mitochondrial genome of Gossypium hirsutum and evolutionary analysis of higher plant mitochondrial genomes. PLoS ONE 2013, 8, e69476. [Google Scholar] [CrossRef]
- Cheng, Y.; He, X.X.; Priyadarshani, S.V.G.N.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.Z.; Zhou, Q.; Luo, Z.Q.; Deng, F.; et al. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef]
- Loizeau, P.A.; Barriea, G.; Manen, J.F.; Broennimann, O. Towards an understanding of the distribution of Ilex, L. (Aquifoliaceae) on a world-wide scale. Biol. Skr. 2005, 55, 501–520. Available online: https://www.researchgate.net/publication/237551705 (accessed on 14 October 2023).
- Yao, X.; Song, Y.; Yang, J.B.; Tan, Y.H.; Corlett, R.T. Phylogeny and biogeography of the hollies (Ilex L., Aquifoliaceae). J. Syst. Evol. 2021, 59, 73–82. [Google Scholar] [CrossRef]
- Zhou, P.; Zhu, Y.Y.; Liu, B.; Li, F.; Huang, J.; Zhang, M. Geographical distribution pattern of species diversity of the genus Ilex in China. J. Cent. South Univ. For. Technol. 2022, 42, 126–132. [Google Scholar] [CrossRef]
- Zhang, C.X.; Lin, C.Z.; Xiong, T.Q.; Zhu, C.C.; Yang, J.Y.; Zhao, Z.X. New triter pene saponins from the root of Ilex pubescens. Fitoterapia 2010, 81, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.; Xie, M.H.; Chen, G.J.; Dai, Z.Q.; Hu, B.; Zeng, X.X.; Sun, Y. Anti-inflammatory effects of dicaffeoylquinic acids from Ilex kudingcha on lipopolysaccharide-treated RAW264.7 macrophages and potential mechanisms. Food Chem. Toxicol. 2019, 126, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Q.; Hao, Y.F.; Xu, Y. Characterization of the complete mitochondrial genome of Ilex pubescens. Mitochondrial DNA Part B 2019, 4, 2003–2004. [Google Scholar] [CrossRef]
- Zhou, P.; Zhang, Q.; Li, F.; Huang, J.; Zhang, M. Assembly and comparative analysis of the complete mitochondrial genome of Ilex metabaptista (aquifoliaceae), a chinese endemic species with a narrow distribution. BMC Plant Biol. 2023, 23, 393. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Lu, Z.Q.; Song, Y.; Hu, X.D.; Corlett, R.T. A chromosome-scale genome assembly for the holly (Ilex polyneura) provides insights into genomic adaptations to elevation in Southwest China. Hortic. Res. 2022, 9, uhab049. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Sebastian, B.; Thomas, T.; Thomas, M.; Uwe, S.; Martin, M. MISAweb: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats fnder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, W.C.; Zhang, Y.D.; Xu, Y.H. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [PubMed]
- Edera, A.A.; Small, I.; Milone, D.H.; Sanchez-Puerta, M.V. Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 2021, 136, 104682. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Alexander, K.; Rowan, B.A.; Dean, L.; Lidija, B.; Eric, S.M.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef]
- Chevigny, N.; Schatz-Daas, D.; Lotfi, F.; Gualberto, J.M. DNA repair and the stability of the plant mitochondrial genome. Int. J. Mol. Sci. 2020, 21, 328. [Google Scholar] [CrossRef] [PubMed]
- Wynn, E.L.; Christensen, A.C. Repeats of unusual size in plant mitochondrial genomes: Identification, incidence and evolution. Genes Genomes Genet. 2019, 9, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Gui, S.T.; Wu, Z.H.; Zhang, H.Y.; Zheng, Y.Z.; Zhu, Z.X.; Liang, D.Q.; Ding, Y. The mitochondrial genome map of Nelumbo nucifera reveals ancient evolutionary features. Sci. Rep. 2016, 6, 30158. [Google Scholar] [CrossRef]
- Liao, X.F.; Zhao, Y.H.; Kong, X.J.; Khan, A.; Zhou, B.J.; Liu, D.M.; Kashif, M.H.; Chen, P.; Wang, H.; Zhou, R.Y. Complete sequence of kenaf (Hibiscus cannabinus) mitochondrial genome and comparative analysis with the mitochondrial genomes of other plants. Sci. Rep. 2018, 8, 12714. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.W.; Qu, Y.S.; Hou, J.; Wu, K.; Ye, N.; Yin, T.M. Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front. Plant Sci. 2022, 13, 914635. [Google Scholar] [CrossRef]
- Han, F.h.; Qu, Y.s.; Chen, Y.c.; Xu, L.a.; Bi, C.W. Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 2022, 13, 1031769. [Google Scholar] [CrossRef]
- Tang, D.F.; Huang, S.H.; Quan, C.Q.; Huang, Y.; Miao, J.H.; Wei, F. Mitochondrial genome characteristics and phylogenetic analysis of the medicinal and edible plant Mesona chinensis Benth. Frontiers in Genetics. Front. Genet. 2023, 13, 1056389. [Google Scholar] [CrossRef]
- Cao, Y.; Yin, D.; Pang, B.; Li, H.B.; Liu, Q.; Zhai, Y.F.; Ma, N.; Shen, H.J.; Jia, Q.J.; Wang, D.K. Assembly and phylogenetic analysis of the mitochondrial genome of endangered medicinal plant Huperzia crispata. Funct. Integr. Genom. 2023, 23, 295. [Google Scholar] [CrossRef]
- Christensen, A.C. Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol. Evol. 2013, 5, 1079–1086. [Google Scholar] [CrossRef]
- Qiao, Y.G.; Zhang, X.R.; Li, Z.; Song, Y.; Sun, Z. Assembly and comparative analysis of the complete mitochondrial genome of Bupleurum chinense DC. BMC Genom. 2022, 23, 664. [Google Scholar] [CrossRef]
- Bi, C.W.; Paterson, A.H.; Wang, X.L.; Xu, Y.Q.; Wu, D.Y.; Qu, Y.S.; Jiang, A.; Ye, Q.L.; Ye, N. Analysis of the complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. BioMed Res. Int. 2016, 2016, 5040598. [Google Scholar] [CrossRef] [PubMed]
- Unseld, M.; Marienfeld, J.R.; Brandt, P.; Brennicke, A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 1997, 15, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome:frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genom. 2002, 268, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.Y.; Wang, Y.X.; Li, S.S.; Wen, J.; Zhu, L.; Yan, K.Y.; Du, Y.M.; Ren, J.; Li, S.X.; Chen, Z.; et al. Assembly and comparative analysis of the first complete mitochondrial genome of Acer truncatum Bunge: A woody oil-tree species producing nervonic acid. BMC Plant Biol. 2022, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Qu, K.; Yuan, Y.C.; Zhao, Z.H.; Chen, Y.; Han, B.; Li, W.; Kassaby, Y.A.; Yin, Y.Y.; Xie, X.M.; et al. Complete sequence and comparative analysis of the mitochondrial genome of the rare and endangered Clematis acerifolia, the first clematis mitogenome to provide new insights into the phylogenetic evolutionary status of the genus. Front. Genet. 2023, 13, 1050040. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.L.; Palmer, J.D. Fine-scale mergers of chloroplast and mitochondrial genes create functional, transcompartmentally chimeric mitochondrial genes. Proc. Natl. Acad. Sci. USA 2009, 106, 16728–16733. [Google Scholar] [CrossRef] [PubMed]
- Goremykin, V.V.; Salamini, F.; Velasco, R.; Viola, R. Mitochondrial DNA of Vitis vinifera and the issue of rampant horizontal gene transfer. Mol. Biol. Evol. 2008, 26, 99–110. [Google Scholar] [CrossRef]
- Lei, B.; Li, S.; Liu, G.; Wang, Y.; Su, A.; Hua, J. Evolutionary analysis of mitochondrial genomes in higher plants. Mol. Plant Breed. 2012, 10, 490–500. [Google Scholar]
- Liu, D.; Guo, H.L.; Zhu, J.L.; Qu, K.; Chen, Y.; Guo, Y.T.; Ding, P.; Yang, H.P.; Xu, T.; Jing, Q.; et al. Complex physical structure of complete mitochondrial genome of Quercus acutissima (Fagaceae): A significant energy plant. Genes 2022, 13, 1321. [Google Scholar] [CrossRef]
- Hepburn, N.J.; Schmidt, D.W.; Mower, J.P. Loss of two introns from the magnolia tripetala mitochondrial cox2 gene implicates horizontal gene transfer and gene conversion as a novel mechanism of intron loss. Mol. Biol. Evol. 2012, 29, 3111–3120. [Google Scholar] [CrossRef]
- Yang, T.Z.; Xu, G.L.; Gu, B.N.; Shi, Y.M.; Mzuka, H.L.; Shen, H.D. The complete mitochondrial genome sequences of the Philomycus bilineatus (Stylommatophora: Philomycidae) and phylogenetic analysis. Genes 2019, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- McLay, T.G.B.; Fowler, R.M.; Fahey, P.S.; Murphy, D.J.; Udovicic, F.; Cantrill, D.J.; Bayly, M.J. Phylogenomics reveals extreme gene tree discordance in a lineage of dominant trees: Hybridization, introgression, and incomplete lineage sorting blur deep evolutionary relationships despite clear species groupings in Eucalyptus subgenus Eudesmia. Mol. Phylogenet. Evol. 2023, 187, 107869. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.T.; Hou, Z.Y.; Chao Li, C.; Yang, J.P.; Niu, Z.T.; Xue, Q.Y.; Liu, W.; Ding, X.Y. Rapid structural evolution of Dendrobium mitogenomes and mito-nuclear phylogeny discordances in Dendrobium (Orchidaceae). J. Syst. Evol. 2022, 61, 790–805. [Google Scholar] [CrossRef]
- Ford, A.G.P.; Bullen, T.R.; Pang, L.; Genner, M.J.; Bills, R.; Flouri, T.; Ngatunga, B.P.; Rüber, L.; Schliewen, U.K.; Seehausen, O.; et al. Molecular phylogeny of Oreochromis (Cichlidae: Oreochromini) reveals mito-nuclear discordance and multiple colonisation of adverse aquatic environments. Mol. Phylogenet. Evol. 2019, 136, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Knie, N.; Polsakiewicz, M.; Knoop, V. Horizontal gene transfer of chlamydial-like tRNA genes into early vascular plant mitochondria. Mol. Biol. Evol. 2015, 32, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.B.; Ren, C.; Kwak, M.; Richard, G.J.; Hodel, R.G.J.; Chao Xu, C.; Jian He, J.; Zhou, W.B.; Huang, C.H.; Hong Ma, H.; et al. Phylogenomic conflict analyses in the apple genus Malus s.l. reveal widespread hybridization and allopolyploidy driving diversification, with insights into the complex biogeographic history in the Northern Hemisphere. J. Integr. Plant Biol. 2022, 64, 1020–1043. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Du, Y.X.; Folk, R.A.; Wang, S.Y.; Li, P. Plastome Evolution in Saxifragaceae and Multiple Plastid Capture Events Involving Heuchera and Tiarella. Front. Plant Sci. 2020, 11, 361. [Google Scholar] [CrossRef]
- Yin, H.; Akimoto, M.; Kaewcheenchai, R.; Sotowa, M.; Ishikawa, R. Inconsistent diversities between nuclear and plastid genomes of AA genome species in the genus Oryza. Genes Genet. Syst. 2015, 90, 269–281. [Google Scholar] [CrossRef]
- Lin, H.Y.; Hao, Y.J.; Li, J.H.; Fu, C.X.; Soltis, P.S.; Soltis, D.E.; Zhao, Y.P. Phylogenomic conflict resulting from ancient introgression following species diversification in Stewartia s.l. (Theaceae). Mol. Phylogenetics Evol. 2019, 135, 1–11. [Google Scholar] [CrossRef]
- Morales-Briones, D.F.; Kadereit, G.; Tefarikis, D.T.; Moore, M.J.; Smith, S.A.; Brockington, S.F.; Timoneda, A.; Yim, W.C.; Cushman, J.C.; Yang, Y. Disentangling sources of gene tree discordance in phylogenomic datasets: Testing ancient Hybridizations in Amaranthaceae s.l. Syst. Biol. 2021, 70, 219–235. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | A % | C % | G % | T % | GC % | Size (bp) | Proportion in Genome (%) |
|---|---|---|---|---|---|---|---|
| Whole genome | 27.20 | 22.54 | 22.99 | 27.27 | 45.53 | 539,461 | 100 |
| Protein-coding genes | 26.40 | 21.40 | 21.81 | 30.38 | 43.21 | 32,767 | 6.07 |
| tRNA genes a | 26.19 | 27.45 | 23.18 | 23.18 | 50.63 | 1428 | 0.26 |
| rRNA genes a | 26.14 | 22.66 | 29.19 | 22.01 | 51.85 | 5248 | 0.97 |
| Non-coding regions | 27.27 | 22.60 | 23.00 | 27.13 | 45.60 | 500,018 | 92.69 |
| Motif Type | Number of Repeats | Total | Proportion (%) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 15 | 16 | 18 | 20 | |||
| Monomer | 21 | 6 | 6 | 3 | 1 | 2 | 1 | 40 | 23.53 | ||||||||
| Dimer | 27 | 8 | 2 | 2 | 39 | 22.94 | |||||||||||
| Trimer | 11 | 2 | 13 | 7.65 | |||||||||||||
| Tetramer | 59 | 2 | 1 | 62 | 36.47 | ||||||||||||
| Pentamer | 11 | 1 | 1 | 2 | 15 | 8.82 | |||||||||||
| Hexamer | 1 | 1 | 0.59 | ||||||||||||||
| Total | 71 | 14 | 30 | 8 | 2 | 1 | 2 | 21 | 6 | 6 | 3 | 1 | 2 | 2 | 1 | 170 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Sun, N.; Shi, W.; Ma, Q.; Sun, L.; Hao, M.; Bi, C.; Li, S. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Ilex macrocarpa. Forests 2023, 14, 2372. https://doi.org/10.3390/f14122372
Wang Y, Sun N, Shi W, Ma Q, Sun L, Hao M, Bi C, Li S. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Ilex macrocarpa. Forests. 2023; 14(12):2372. https://doi.org/10.3390/f14122372
Chicago/Turabian StyleWang, Yuxiao, Ning Sun, Wenxi Shi, Qiuyue Ma, Liyong Sun, Mingzhuo Hao, Changwei Bi, and Shuxian Li. 2023. "Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Ilex macrocarpa" Forests 14, no. 12: 2372. https://doi.org/10.3390/f14122372