
Identification of lncRNAs and Their Regulatory Network Involved in Oil Biosynthesis in Developing Seeds of Yellowhorn (Xanthoceras sorbifolium)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Oil Content Analysis
2.3. RNA Extraction and Strand-Specific Library Construction
2.4. Identification of lncRNAs and mRNAs
2.5. Identification of Differentially Expressed lncRNAs (DELs) and mRNAs (DEGs)
2.6. LncRNA Target mRNA Prediction
2.7. Small RNA Sequencing
2.8. Identification of Known and Novel miRNAs and Prediction of Their Targets
2.9. Analysis of Differentially Expressed miRNAs (DEM)
2.10. GO and KEGG Enrichment Analysis
2.11. qPCR Assay
2.12. Statistical Analysis
3. Results
3.1. Dynamic Changes of Oil Content during Seed Development
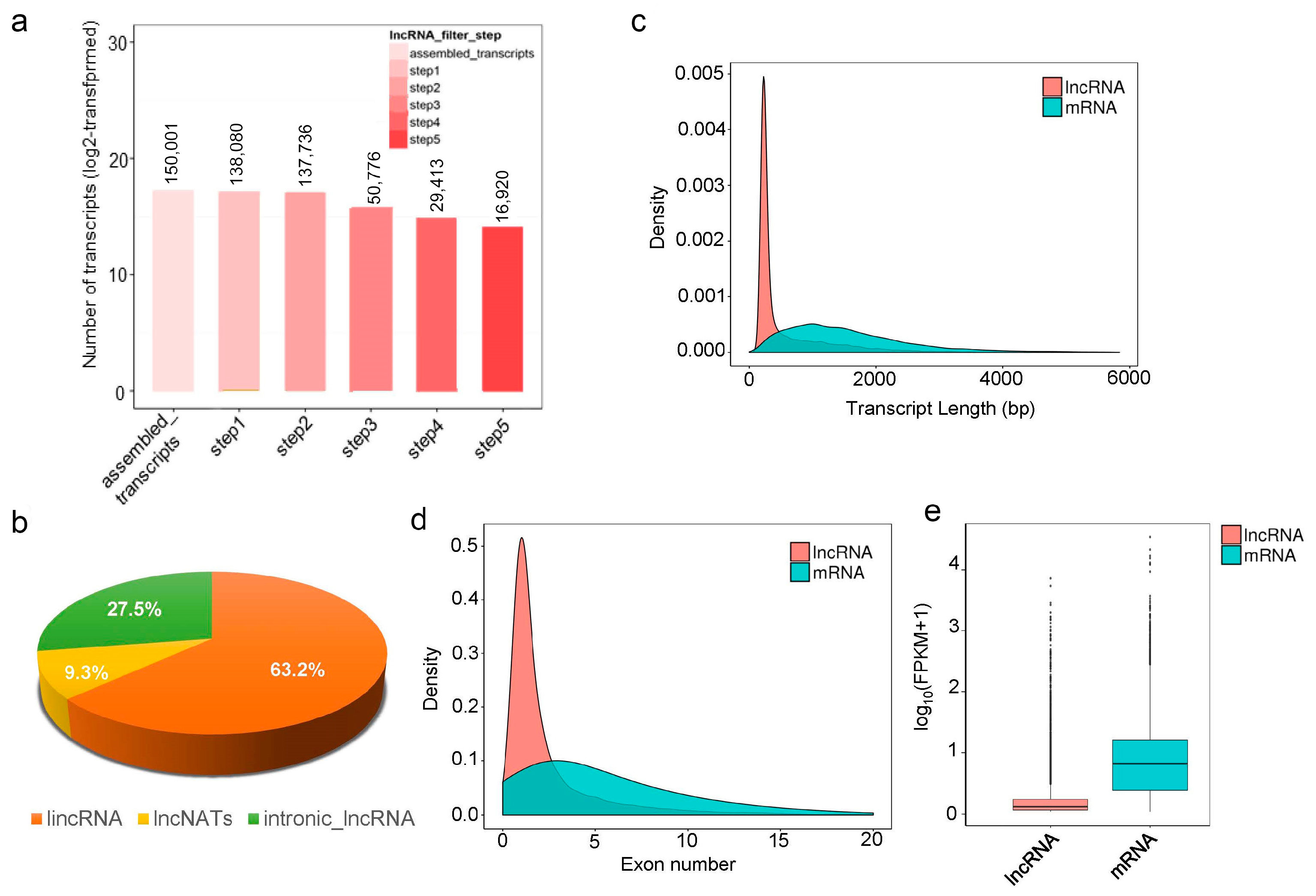
3.2. Transcriptome Analysis during Seed Development
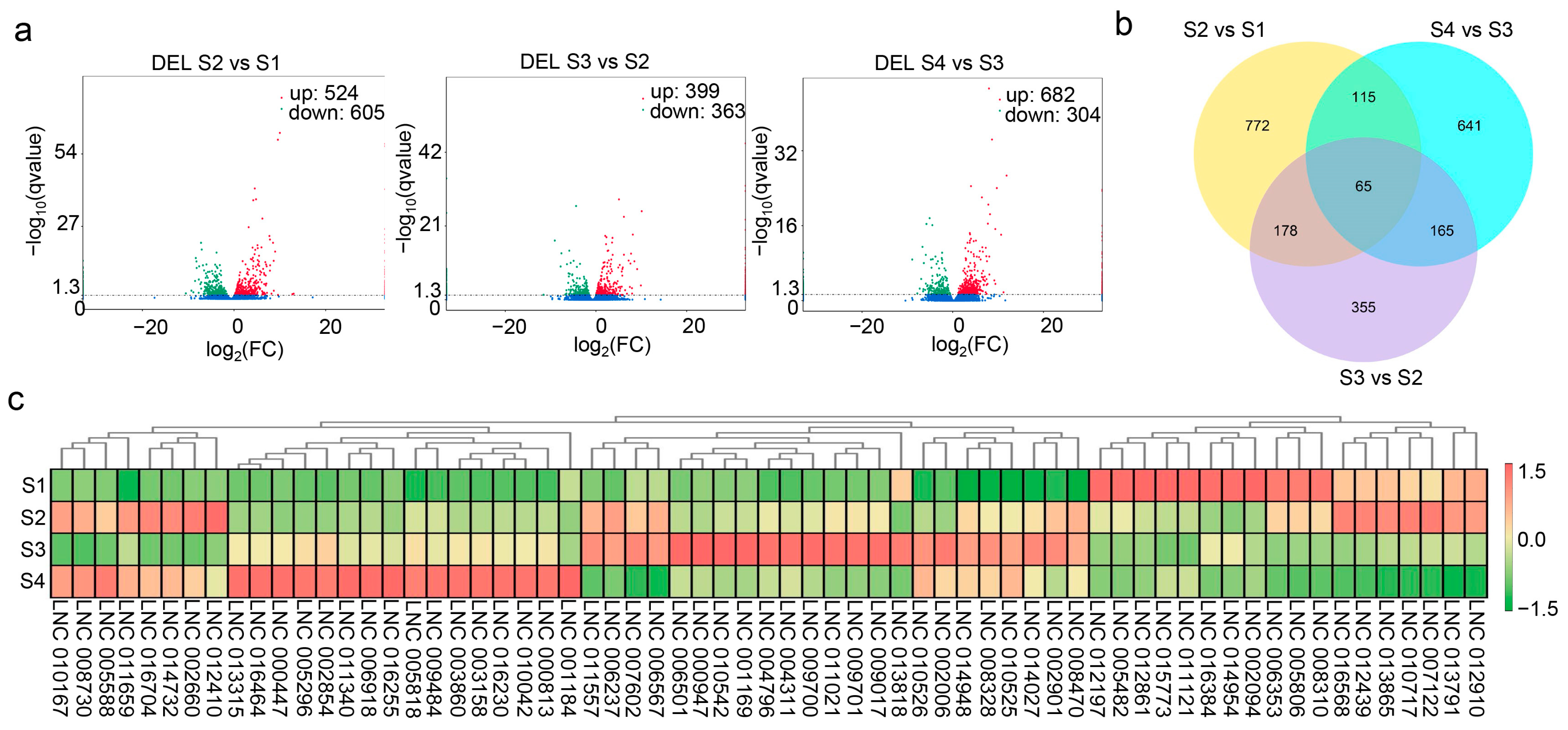
3.3. Differential Expression Profiles of lncRNAs
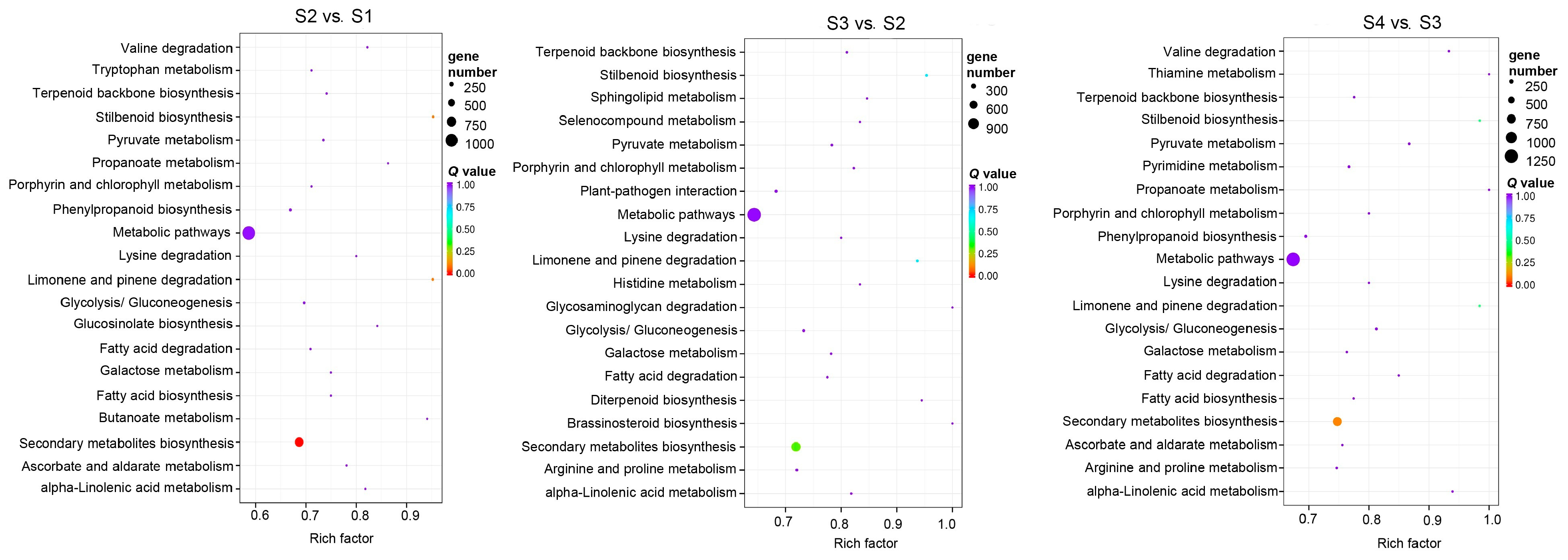
3.4. GO Enrichment and KEGG Enrichment Analysis of DELs
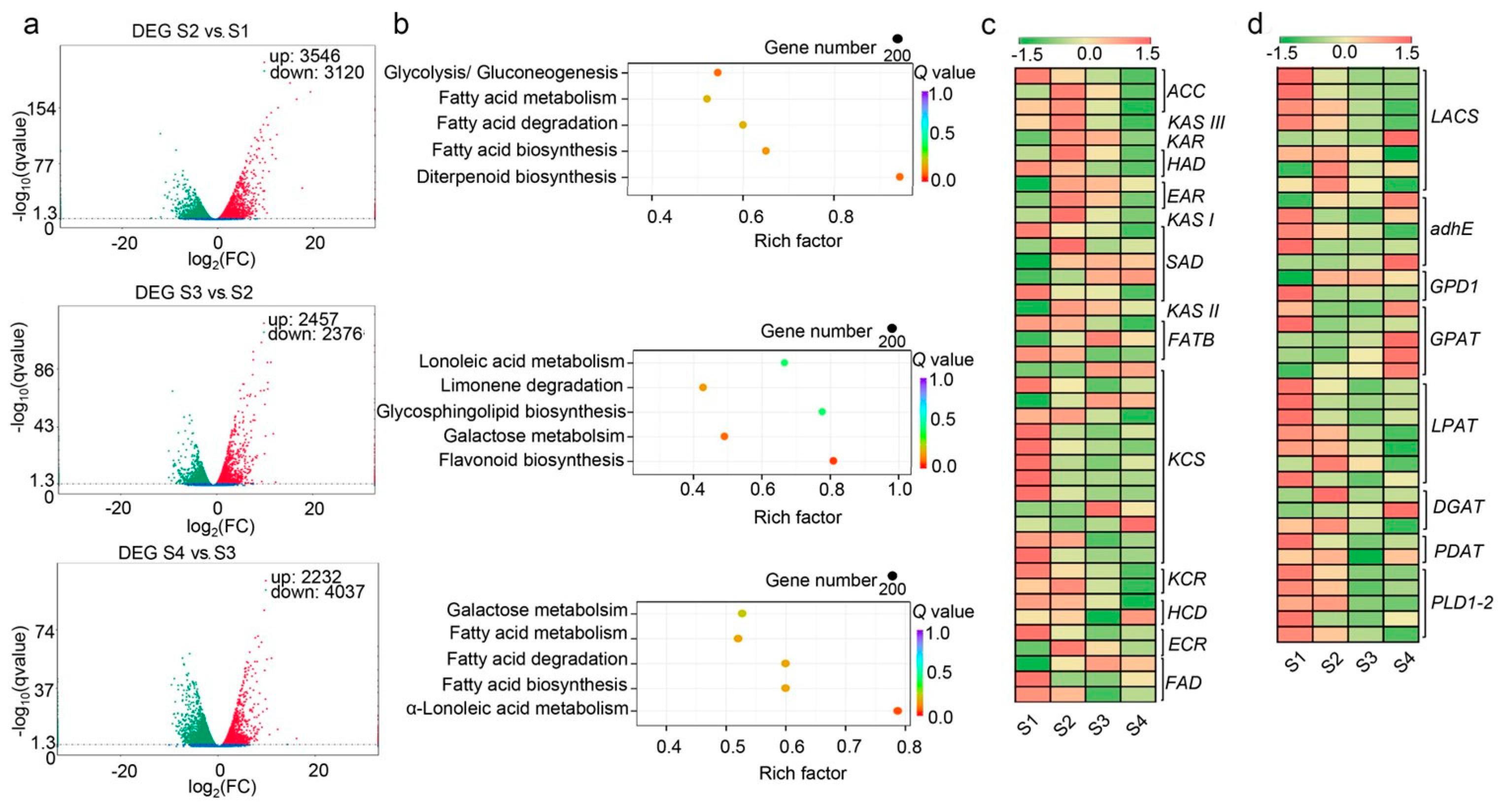
3.5. DEGs Involved in FA and TAG Biosynthesis
3.6. DEL–DEG Pairs Related to FA and TAG Biosynthesis
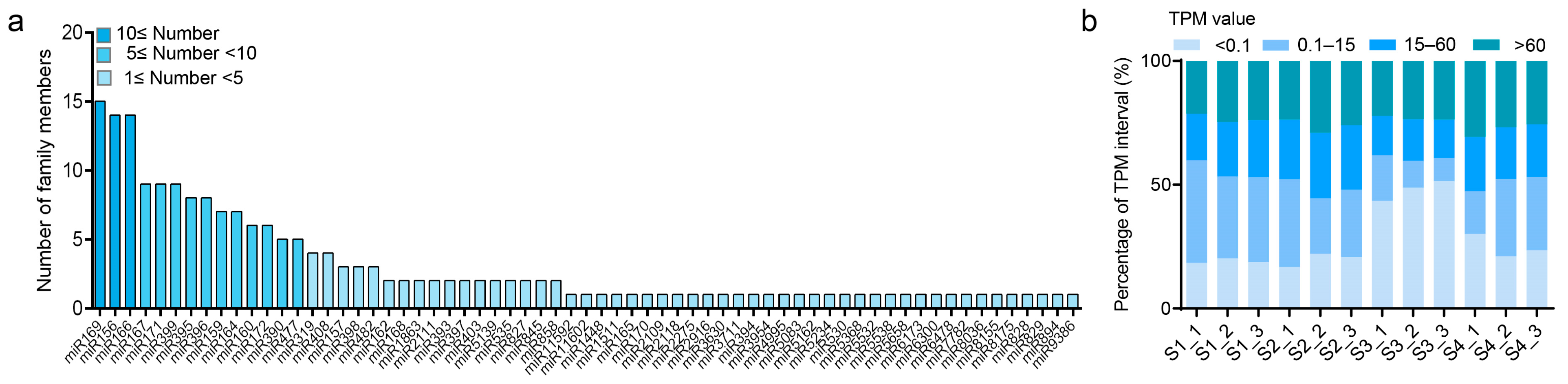
3.7. Small RNA Sequencing Profile
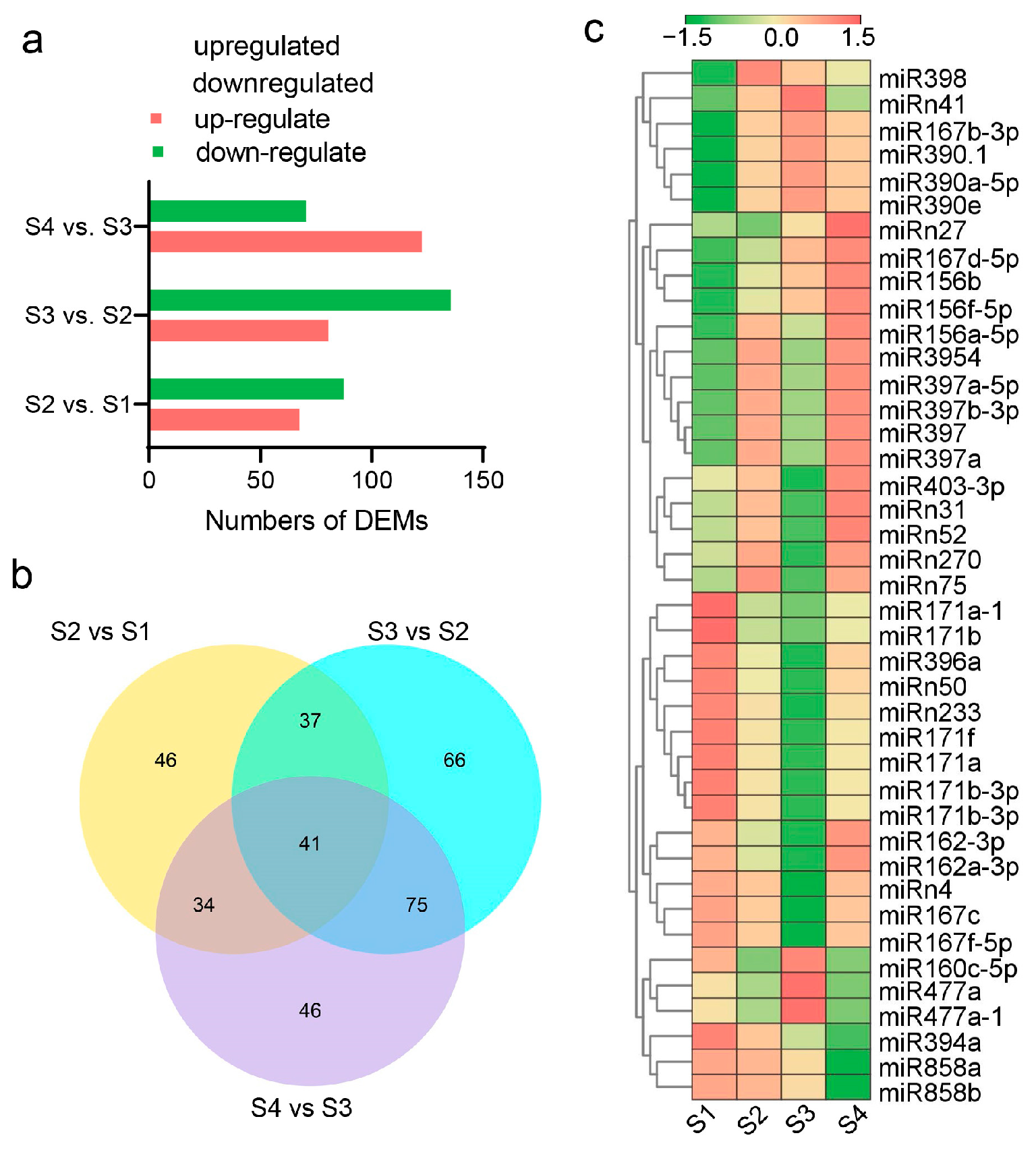
3.8. Different Expression Profiles of miRNAs
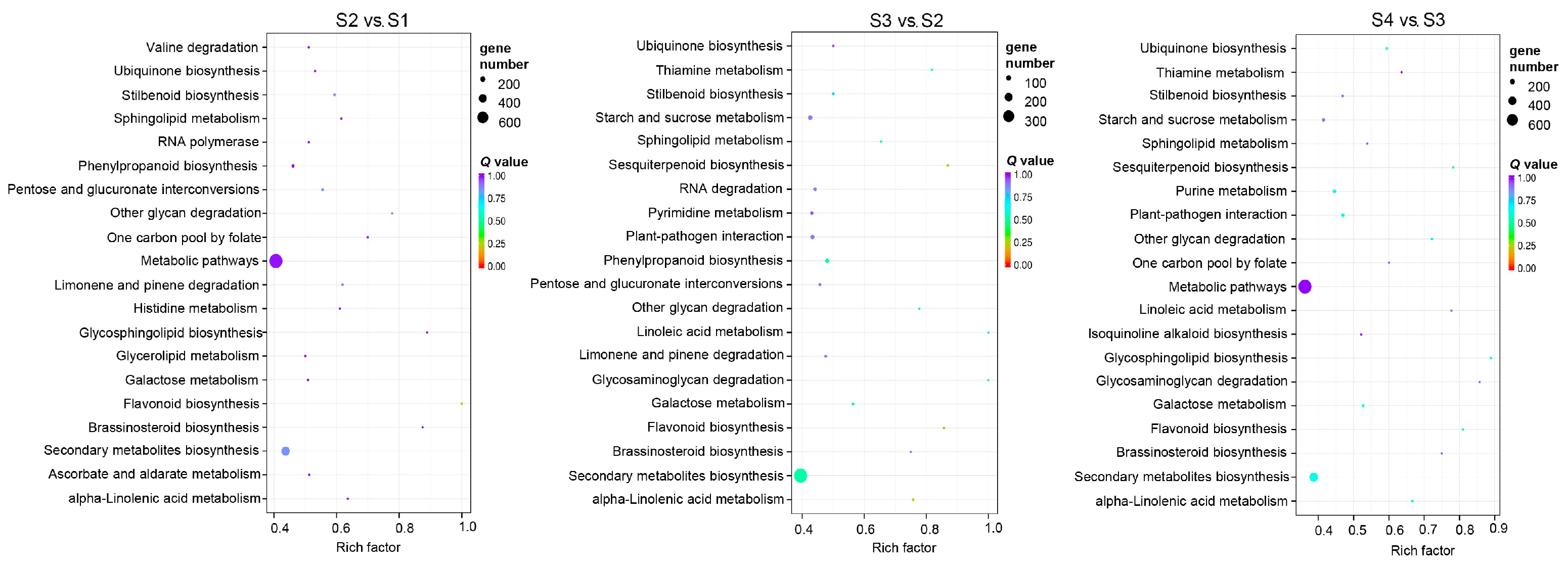
3.9. GO Enrichment and KEGG Enrichment Analysis of DEMs
3.10. DEM–DEG Pairs in the Context of FA and TAG Synthesis Pathways
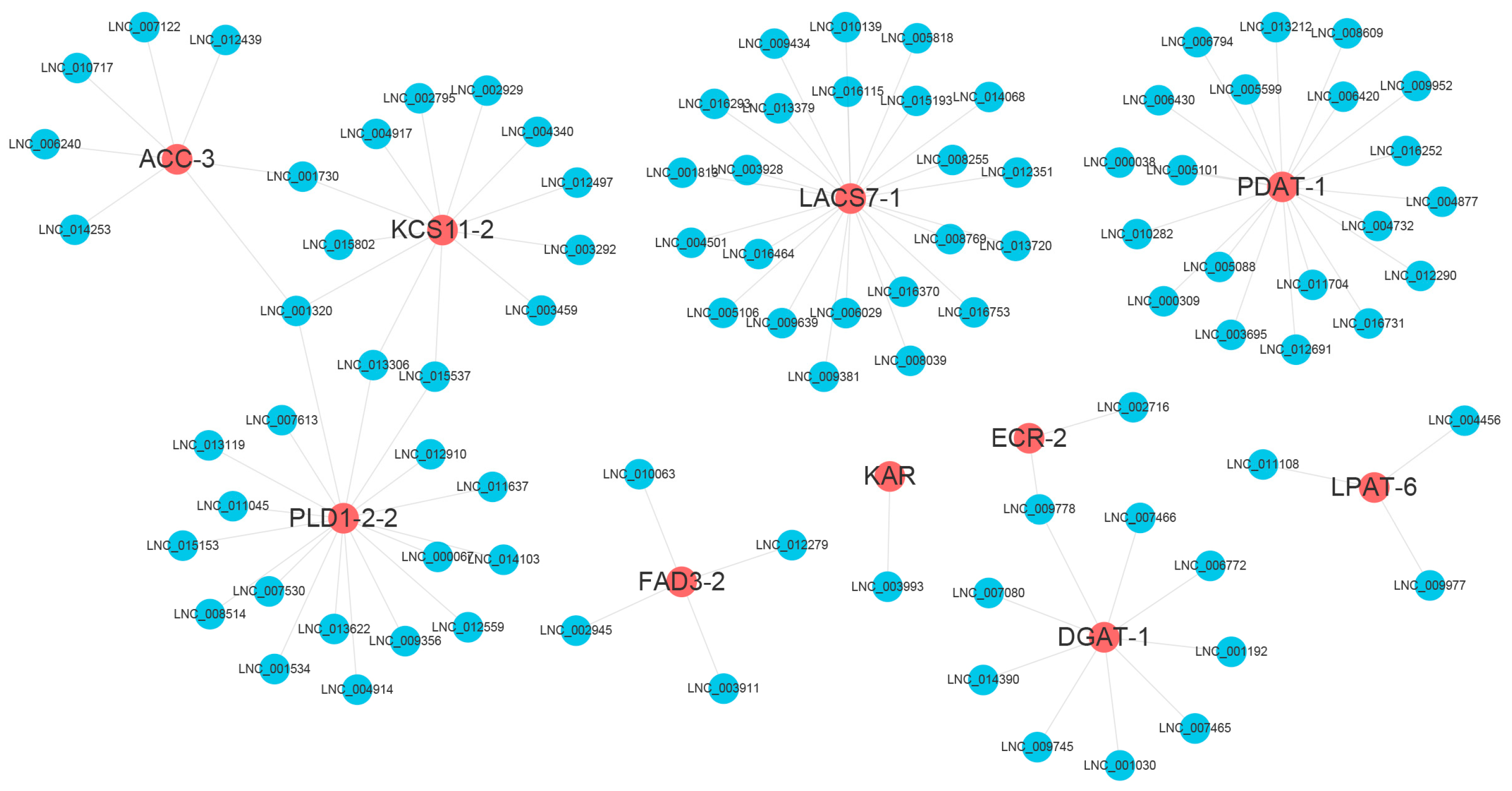
3.11. DEL–DEM–DEG Network Involved in FA and TAG Synthesis
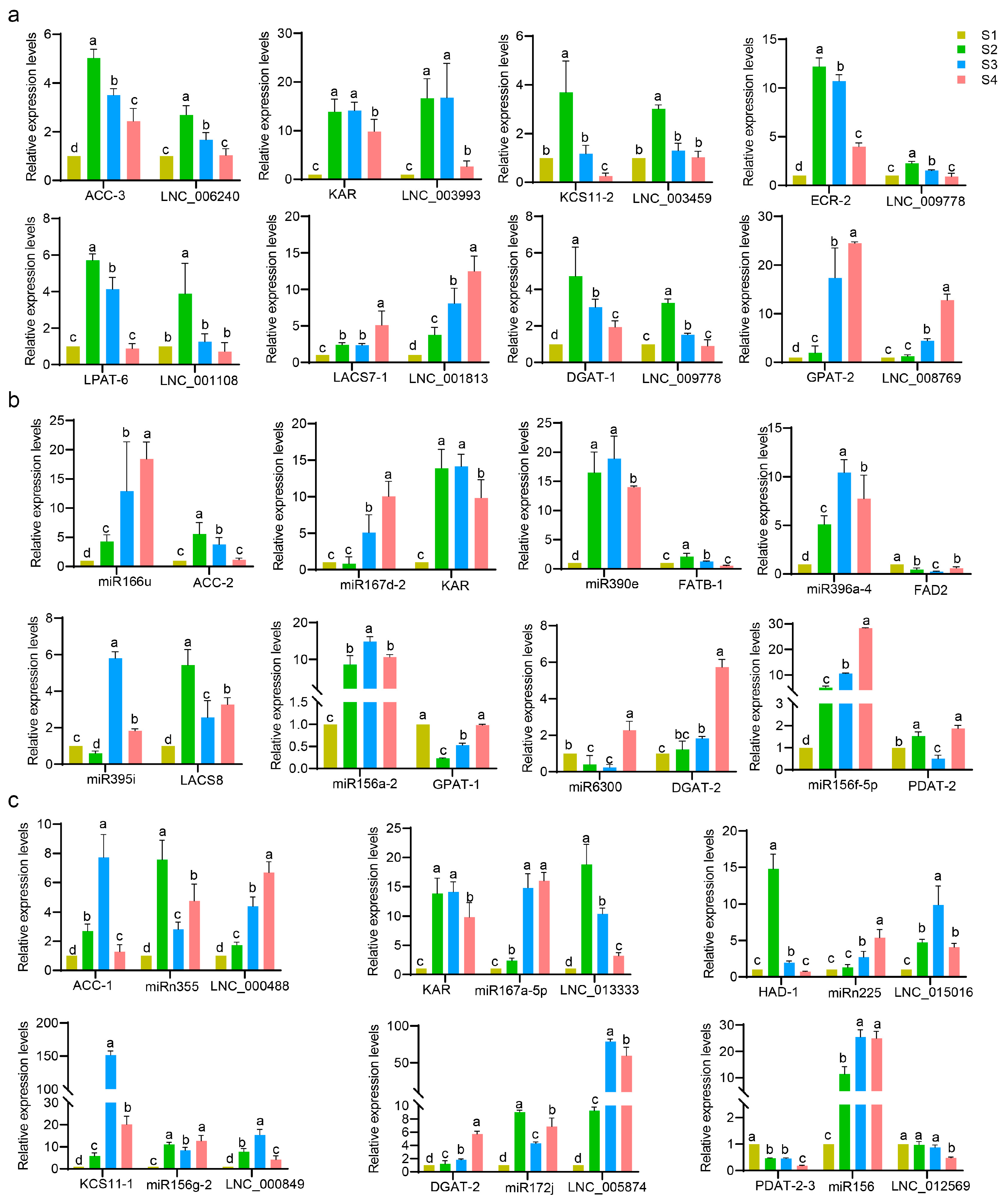
3.12. Verification of DEL–DEG Pairs, DEM–DEG Pairs and DEL–DEM–DEG Modules by Quantitative Assay
4. Discussion
4.1. LncRNAs Identified in Developing Seeds of Yellowhorn
4.2. Key lncRNAs and miRNAs Involved in TAG Assembly of Yellowhorn
4.3. Key lncRNAs and miRNAs Involved in the FA Biosynthesis Pathway
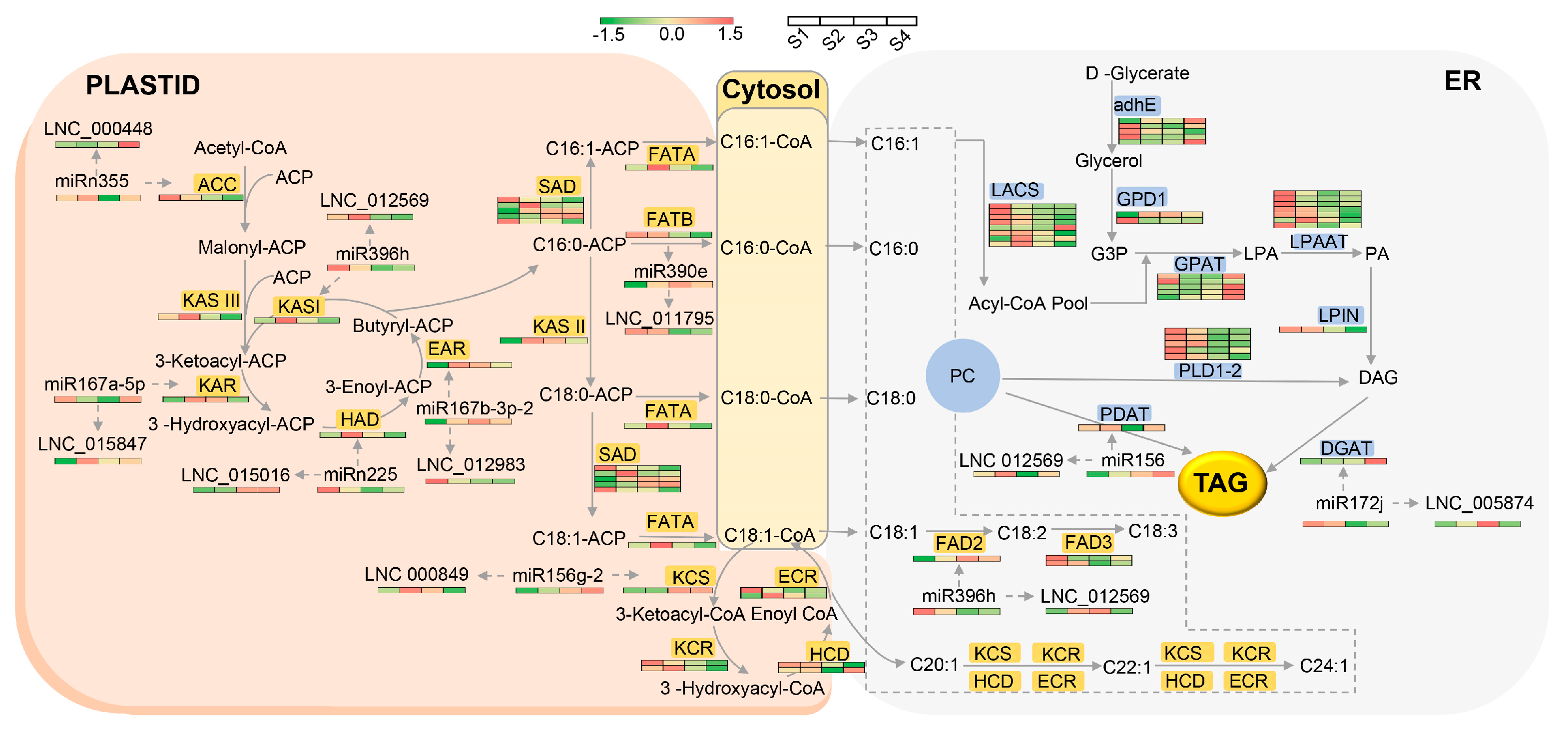
4.4. Working Model of the lncRNA–miRNA–mRNA Modules in FA and TAG Synthesis Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Venegas-Calerón, M.; Ruíz-Méndez, M.V.; Martínez-Force, E.; Garcés, R.; Salas, J.J. Characterization of Xanthoceras sorbifolium Bunge seeds: Lipids, proteins and saponins content. Ind. Crops Prod. 2017, 109, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Ruan, C.J.; Yan, R.; Wang, B.X.; Mopper, S.; Guan, W.K.; Zhang, J. The importance of yellow horn (Xanthoceras sorbifolia) for restoration of arid habitats and production of bioactive seed oils. Ecol. Eng. 2017, 99, 504–512. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, X.; Wang, M.; Bi, Q.; Cui, Y.; Wang, L. Transcriptome and physiological analyses provide insights into the leaf epicuticular wax accumulation mechanism in yellowhorn. Hortic. Res. 2021, 8, 134. [Google Scholar] [CrossRef]
- Bates, P.D.; Stymne, S.; Ohlrogge, J. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Okuley, J.; Lightner, J.; Feldmann, K.; Yadav, N.; Lark, E.; Browse, J. Arabidopsis FAD2 gene encodes the enzyme that is essential for polyunsaturated lipid synthesis. Plant Cell 1994, 6, 147–158. [Google Scholar]
- Vrinten, P.; Hu, Z.; Munchinsky, M.A.; Rowland, G.; Qiu, X. Two FAD3 desaturase genes control the level of linolenic acid in flax seed. Plant Physiol. 2005, 139, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Fillet, S.; Ronchel, C.; Callejo, C.; Fajardo, M.J.; Moralejo, H.; Adrio, J.L. Engineering Rhodosporidium toruloides for the production of very long-chain monounsaturated fatty acid-rich oils. Appl. Microbiol. Biot. 2017, 101, 7271–7280. [Google Scholar] [CrossRef]
- Correa, S.M.; Fernie, A.R.; Nikoloski, Z.; Brotman, Y. Towards model-driven characterization and manipulation of plant lipid metabolism. Prog. Lipid Res. 2020, 80, 101051. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.D.; Li, S.S.; Shu, Q.Y.; Gu, Z.Y.; Wu, Q.; Feng, C.Y.; Xu, W.Z.; Wang, L.S. Identification of microRNAs and long non-coding RNAs involved in fatty acid biosynthesis in tree peony seeds. Gene 2018, 666, 72–82. [Google Scholar] [CrossRef]
- Zhang, C.C.; Ren, H.D.; Yao, X.H.; Wang, K.L.; Chang, J. Full-length transcriptome analysis of pecan (Carya illinoinensis) kernels. G3-Genes Genom. Genet. 2021, 11, jkab182. [Google Scholar] [CrossRef]
- Xia, W.; Dou, Y.J.; Liu, R.; Gong, S.F.; Huang, D.Y.; Fan, H.K.; Xiao, Y. Genome-wide discovery and characterization of long noncoding RNAs in African oil palm (Elaeis guineensis Jacq.). PeerJ 2020, 8, e9585. [Google Scholar] [CrossRef]
- Shen, E.; Zhu, X.; Hua, S.; Chen, H.; Ye, C.; Zhou, L.; Liu, Q.; Zhu, Q.H.; Fan, L.; Chen, X. Genome-wide identification of oil biosynthesis-related long non-coding RNAs in allopolyploid Brassica napus. BMC Genom. 2018, 19, 745. [Google Scholar] [CrossRef]
- Chen, J.; Wu, Z.R.; Li, R.Y.; Huang, D.J.; Zhai, W.; Chen, C.H. New insight into LncRNA-mRNA regulatory network associated with lipid biosynthesis using Hi-C data in seeds of tung tree (Vernicia fordii Hemsl.). Ind. Crops Prod. 2021, 164, 113321. [Google Scholar] [CrossRef]
- Ao, Y. Characterization and comparison of flower bud microRNAs from yellow-horn species. Genet. Mol. Res. 2016, 15, gmr.15048899. [Google Scholar] [CrossRef]
- Wang, L.; Ruan, C.J.; Bao, A.M.; Li, H. Small RNA profiling for identification of microRNAs involved in regulation of seed development and lipid biosynthesis in yellowhorn. BMC Plant Biol. 2021, 21, 464. [Google Scholar] [CrossRef]
- Yang, W.; Wang, G.; Li, J.; Bates, P.D.; Wang, X.; Allen, D.K. Phospholipase Dζ Enhances Diacylglycerol Flux into Triacylglycerol. Plant Physiol. 2017, 174, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Nigam, D.; Junaid, A.; Tribhuvan, K.U.; Kumar, K.; Durgesh, K.; Singh, N.K.; Gaikwad, K. Expressivity of the key genes associated with seed and pod development is highly regulated via lncRNAs and miRNAs in Pigeonpea. Sci. Rep. 2019, 9, 18191. [Google Scholar] [CrossRef] [Green Version]
- Bi, Q.; Zhao, Y.; Du, W.; Lu, Y.; Gui, L.; Zheng, Z.; Yu, H.; Cui, Y.; Liu, Z.; Cui, T.; et al. Pseudomolecule-level assembly of the Chinese oil tree yellowhorn (Xanthoceras sorbifolium) genome. GigaScience 2019, 8, giz070. [Google Scholar] [CrossRef]
- Shockey, J.M.; Gidda, S.K.; Chapital, D.C.; Kuan, J.C.; Dhanoa, P.K.; Bland, J.M.; Rothstein, S.J.; Mullen, R.T.; Dyer, J.M. Tung tree DGAT1 and DGAT2 have nonredundant functions in triacylglycerol biosynthesis and are localized to different subdomains of the endoplasmic reticulum. Plant Cell 2006, 18, 2294–2313. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J. and Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lin, X.; Lin, W.G.; Ku, Y.S.; Wong, F.L.; Li, M.W.; Lam, H.M.; Ngai, S.M.; Chan, T.F. Analysis of Soybean Long Non-Coding RNAs Reveals a Subset of Small Peptide-Coding Transcripts. Plant Physiol. 2020, 182, 1359–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.L.; Zhang, X.G.; Traore, S.M.; Xin, Z.Y.; Ning, L.L.; Li, K.; Zhao, K.K.; Li, Z.F.; He, G.H.; Yin, D.M. Genome-wide identification and analysis of long noncoding RNAs (lncRNAs) during seed development in peanut (Arachis hypogaea L.). BMC Plant Biol. 2020, 20, 192. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.Y.; Li, S.; Hu, L.Y.; Zhang, C.L. Genome-wide analysis of long non-coding RNAs (lncRNAs) in two contrasting rapeseed (Brassica napus L.) genotypes subjected to drought stress and re-watering. BMC Plant Biol. 2020, 20, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanaka, T.; Serson, W.; Li, R.Z.; Armstrong, P.; Yu, K.S.; Pfeiffer, T.; Li, X.L.; Hildebrand, D. A Vernonia Diacylglycerol Acyltransferase Can Increase Renewable Oil Production. J. Agric. Food Chem. 2016, 64, 7188–7194. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Peng, F.Y.; Weselake, R.J. Genome-wide analysis of phospholipid:diacylglycerol acyltransferase (PDAT) genes in plants reveals the eudicot-wide PDAT gene expansion and altered selective pressures acting on the core eudicot PDAT paralogs. Plant Physiol. 2015, 167, 887–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.M.; Zhou, Y.; Zhang, J.P.; Ji, F.Y.; Jin, F.; Fan, W.; Pei, D. Transcriptome Analysis of Walnut (Juglans regia L.) Embryos Reveals Key Developmental Stages and Genes Involved in Lipid Biosynthesis and Polyunsaturated Fatty Acid Metabolism. J. Agric. Food Chem. 2021, 69, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Siloto, R.M.P.; Wickramarathna, A.D.; Mietkiewska, E.; Weselake, R.J. Identification of a Pair of Phospholipid:Diacylglycerol Acyltransferases from Developing Flax (Linum usitatissimum L.) Seed Catalyzing the Selective Production of Trilinolenin. J. Biol. Chem. 2013, 288, 24173–24188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Liu, W.; Fan, Y.; Zhou, X.; Tang, X.; Zhang, L. Identification and analysis of tRNA genes provide new insights into oil biosynthesis in tung tree (Vernicia fordii Hemsl.). Ind. Crops Prod. 2019, 137, 74–80. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, G.; Liu, X.; Yu, Z.; Peng, S. Integrated Analysis of Seed microRNA and mRNA Transcriptome Reveals Important Functional Genes and microRNA-Targets in the Process of Walnut (Juglans regia) Seed Oil Accumulation. Int. J. Mol. Sci. 2020, 21, 9093. [Google Scholar] [CrossRef]
- Beaudoin, F.; Wu, X.; Li, F.; Haslam, R.P.; Markham, J.E.; Zheng, H.; Napier, J.A.; Kunst, L. Functional characterization of the Arabidopsis beta-ketoacyl-coenzyme A reductase candidates of the fatty acid elongase. Plant Physiol. 2009, 150, 1174–1191. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.W.; Ma, S.J.; Song, H.; Yang, Z.; Zhao, C.Z.; Taylor, D.; Zhang, M. A 3-ketoacyl-CoA synthase 11 (KCS11) homolog from Malania oleifera synthesizes nervonic acid in plants rich in 11Z-eicosenoic acid. Tree Physiol. 2021, 41, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Hajiahmadi, Z.; Abedi, A.; Wei, H.; Sun, W.B.; Ruan, H.H.; Qiang, Z.G.; Movahedi, A. Identification, evolution, expression, and docking studies of fatty acid desaturase genes in wheat (Triticum aestivum L.). BMC Genom. 2020, 21, 778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Zhang, Y.Y.; Thakur, K.; Zhang, F.; Hu, F.; Zhang, J.G.; Wei, P.C.; Wei, Z.J. Integration of miRNAs, Degradome, and Transcriptome Omics Uncovers a Complex Regulatory Network and Provides Insights Into Lipid and Fatty Acid Synthesis During Sesame Seed Development. Front. Plant Sci. 2021, 12, 709197. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, C.; Liang, Y.; Sun, R.; Gao, L.; Liu, T.; Li, D. Genome-wide association analysis of the lipid and fatty acid metabolism regulatory network in the mesocarp of oil palm (Elaeis guineensis Jacq.) based on small noncoding RNA sequencing. Tree Physiol. 2019, 39, 356–371. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Y.; Ruan, C.; Luan, Y.; Li, J. Identification of lncRNAs and Their Regulatory Network Involved in Oil Biosynthesis in Developing Seeds of Yellowhorn (Xanthoceras sorbifolium). Forests 2023, 14, 407. https://doi.org/10.3390/f14020407
Hong Y, Ruan C, Luan Y, Li J. Identification of lncRNAs and Their Regulatory Network Involved in Oil Biosynthesis in Developing Seeds of Yellowhorn (Xanthoceras sorbifolium). Forests. 2023; 14(2):407. https://doi.org/10.3390/f14020407
Chicago/Turabian StyleHong, Yuhui, Chengjiang Ruan, Yushi Luan, and Jingbin Li. 2023. "Identification of lncRNAs and Their Regulatory Network Involved in Oil Biosynthesis in Developing Seeds of Yellowhorn (Xanthoceras sorbifolium)" Forests 14, no. 2: 407. https://doi.org/10.3390/f14020407
APA StyleHong, Y., Ruan, C., Luan, Y., & Li, J. (2023). Identification of lncRNAs and Their Regulatory Network Involved in Oil Biosynthesis in Developing Seeds of Yellowhorn (Xanthoceras sorbifolium). Forests, 14(2), 407. https://doi.org/10.3390/f14020407