
Comparative Transcriptome Analysis between Embryogenic and Non-Embryogenic Callus of Davidia involucrata


Abstract
1. Introduction
2. Materials and Methods
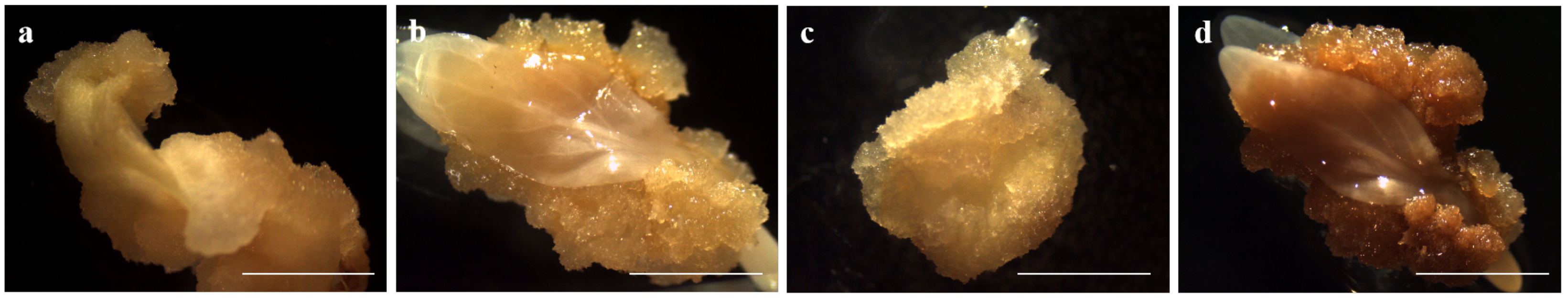
2.1. Materials and Establishment of Somatic Embryogenesis in D. involucrata
2.2. Total RNA Extraction, Library Construction, and Transcriptome Sequencing
2.3. Bioinformatics Analysis of RNA-seq Data
3. Results
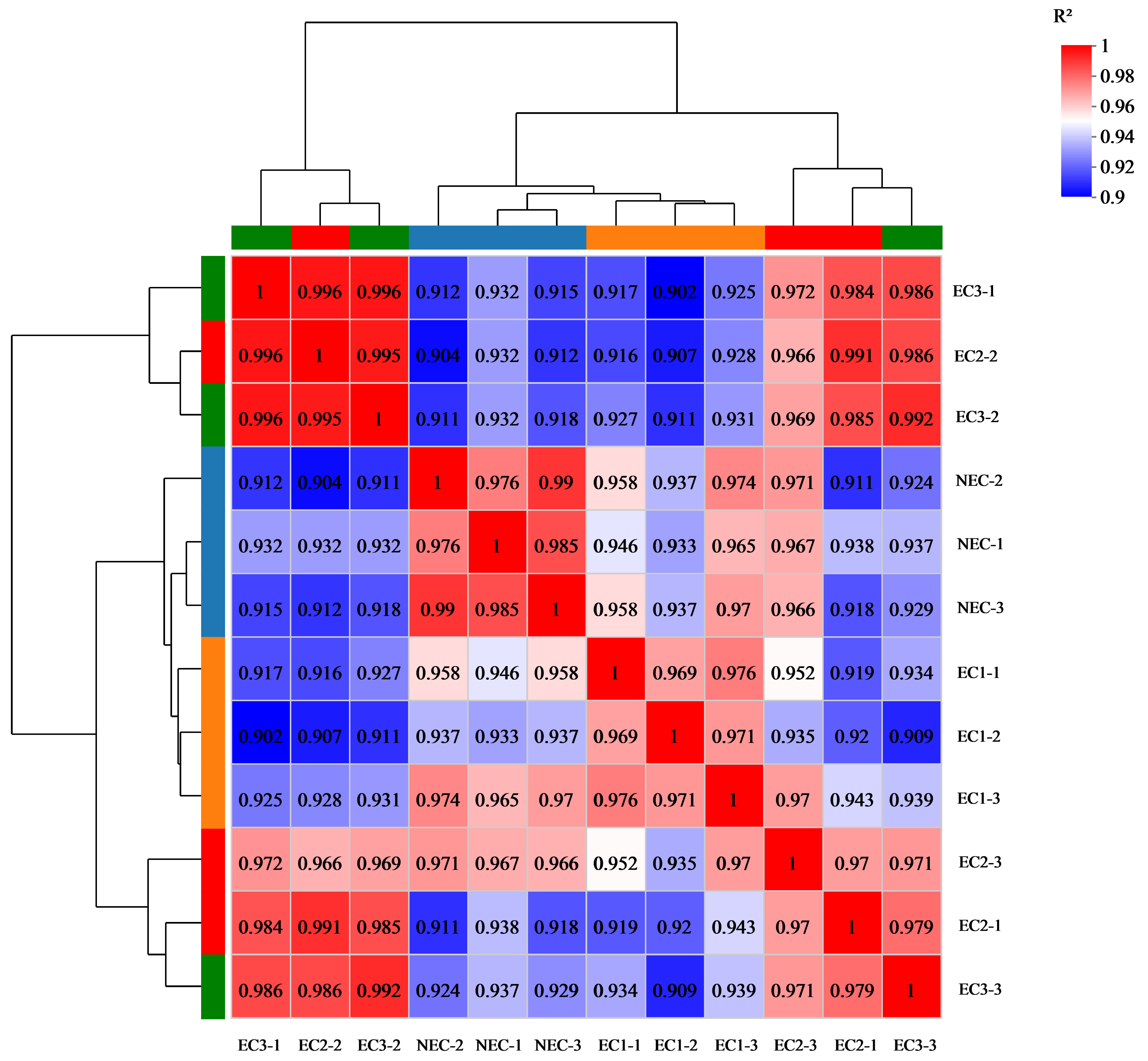
3.1. Transcriptome Sequencing Analysis
3.2. Functional Annotation
3.3. Functional Classification
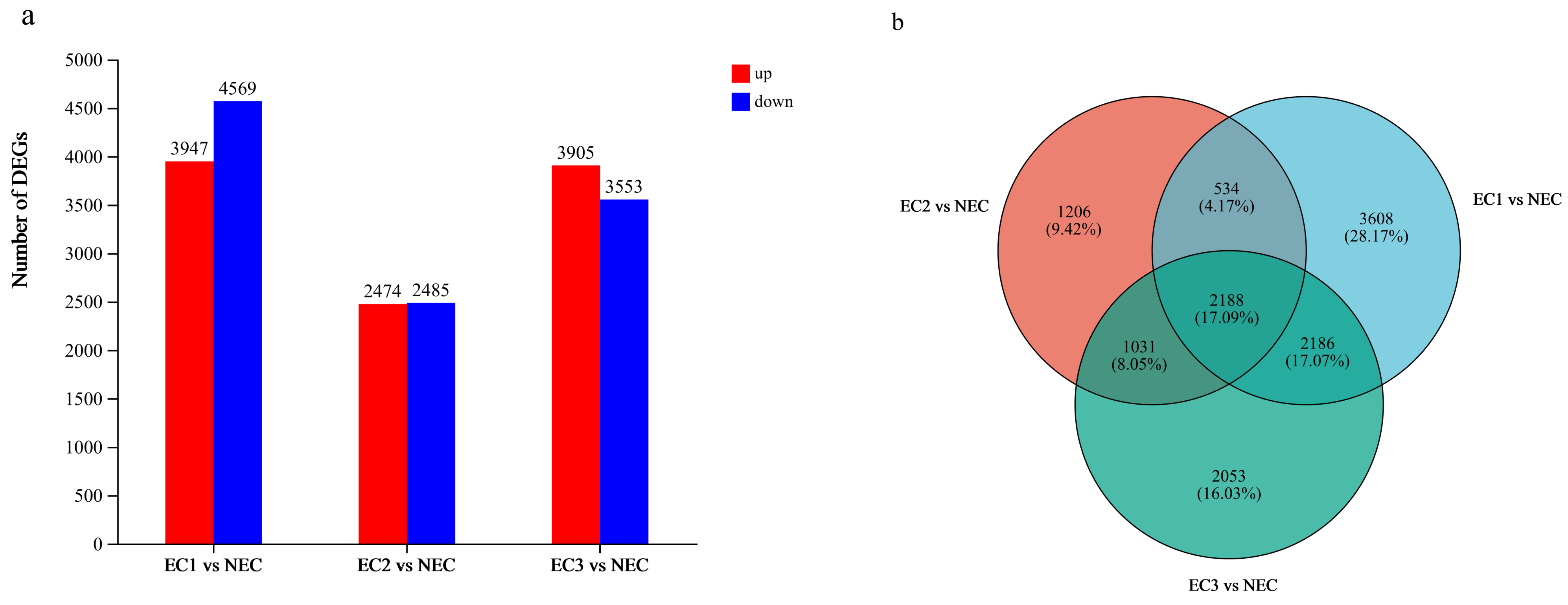
3.4. Analysis of DEGs
3.5. Analysis of Differentially Expressed Transcription Factors
3.6. Expression Analysis of Growth Hormone, Cytokinin, and Stress-Related Genes
3.7. Expression Analysis of Somatic Embryogenesis-Related Genes
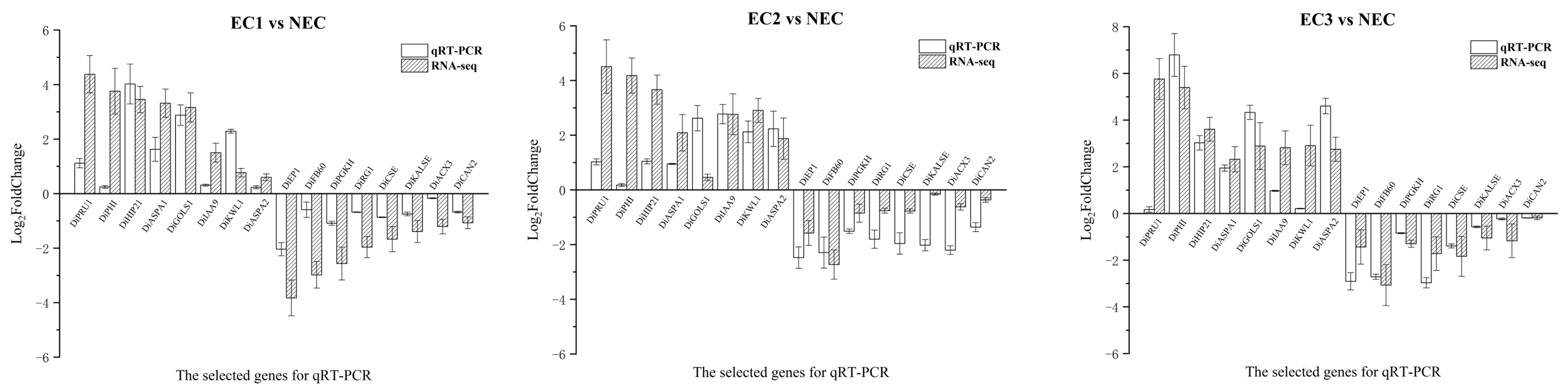
3.8. qRT-PCR
4. Discussion
4.1. The Role of Growth Hormone, Cytokinin, and Stress-Related Genes
4.2. Molecular Regulation of Somatic Embryo-Related Genes in Healing Tissues
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gang, W.; Shan-Heng, H.; Hong-Chang, W.; Yue-Chu, L.; Hong-Bing, D.; Jing-Zhu, Z. Living characteristics of rare and endangered species—Davidia involucrata. J. For. Res. 2004, 15, 39–44. [Google Scholar] [CrossRef]
- Jinsheng, H.; Jie, L.; Weilie, C. The current status of endemic and endangered species Davidia involucrata and the preserving strategies. Biodivers. Sci. 1995, 3, 213. [Google Scholar]
- Jiaxun, Z. Studies on Chinese dovetree propagation and cultivation techniques. J. Beijing For. Univ. 1995, 17, 24–29. [Google Scholar]
- Zimmerman, J.L. Somatic embryogenesis: A model for early development in higher plants. Plant Cell 1993, 5, 1411. [Google Scholar] [CrossRef]
- Ebrahimi, M.; Mokhtari, A.; Amirian, R. A highly efficient method for somatic embryogenesis of Kelussia odorotissima Mozaff., an endangered medicinal plant. Plant Cell Tissue Organ Cult. PCTOC 2018, 132, 99–110. [Google Scholar] [CrossRef]
- Guan, Y.; Li, S.G.; Fan, X.F.; Su, Z.H. Application of somatic embryogenesis in woody plants. Front. Plant Sci. 2016, 7, 938. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Luo, K.; Li, Z.; Yang, Y. A novel method for induction of plant regeneration via somatic embryogenesis. Plant Sci. 2009, 177, 43–48. [Google Scholar] [CrossRef]
- Isah, T. Induction of somatic embryogenesis in woody plants. Acta Physiol. Plant. 2016, 38, 118. [Google Scholar] [CrossRef]
- De Jong, A.J.; Schmidt, E.D.; De Vries, S.C. Early events in higher-plant embryogenesis. Plant Mol. Biol. 1993, 22, 367–377. [Google Scholar] [CrossRef]
- Karami, O.; Aghavaisi, B.; Mahmoudi Pour, A. Molecular aspects of somatic-to-embryogenic transition in plants. J. Chem. Biol. 2009, 2, 177–190. [Google Scholar] [CrossRef]
- Liu, W.; Wang, C.; Shen, X.; Liang, H.; Wang, Y.; He, Z.; Zhang, D.; Chen, F. Comparative transcriptome analysis highlights the hormone effects on somatic embryogenesis in Catalpa bungei. Plant Reprod. 2019, 32, 141–151. [Google Scholar] [CrossRef]
- Kang, H.I.; Lee, C.B.; Kwon, S.H.; Park, J.M.; Kang, K.S.; Shim, D. Comparative transcriptome analysis during developmental stages of direct somatic embryogenesis in Tilia amurensis Rupr. Sci. Rep. 2021, 11, 6359. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Iwase, A.; Rymen, B.; Lambolez, A.; Kojima, M.; Takebayashi, Y.; Heyman, J.; Watanabe, S.; Seo, M.; De Veylder, L.; et al. Wounding triggers callus formation via dynamic hormonal and transcriptional changes. Plant Physiol. 2017, 175, 1158–1174. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Li, P.; Ponnala, L.; Gandotra, N.; Wang, L.; Si, Y.; Tausta, S.L.; Kebrom, T.H.; Provart, N.; Patel, R.; Myers, C.R.; et al. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 2010, 42, 1060. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.N.; Lee, S.A.; Kang, M.J.; Joo, H.J.; Kim, J.A.; Park, E.J. Comparative transcriptome analysis between embryogenic and nonembryogenic callus of Kalopanax septemlobus. For. Sci. Technol. 2020, 16, 145–153. [Google Scholar] [CrossRef]
- Lai, Z.; Lin, Y. Analysis of the global transcriptome of longan (Dimocarpus longan Lour.) embryogenic callus using Illumina paired-end sequencing. BMC Genom. 2013, 14, 561. [Google Scholar] [CrossRef]
- Fehér, A. Somatic embryogenesis—stress-induced remodeling of plant cell fate. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2015, 1849, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, B.; San-José, M.C.; Martinez, M.; Ballester, A.; Vieitez, A. Somatic embryogenesis from stem and leaf explants of Quercus robur L. Plant Cell Rep. 1999, 18, 538–543. [Google Scholar] [CrossRef]
- Altamura, M.M.; Della Rovere, F.; Fattorini, L.; D’Angeli, S.; Falasca, G. Recent advances on genetic and physiological bases of in vitro somatic embryo formation. Vitr. Embryog. High. Plants 2016, 1359, 47–85. [Google Scholar]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Huang, Y.; Chen, J.; Benesty, J.; Benesty, J.; Chen, J.; Huang, Y.; Cohen, I. Pearson correlation coefficient. In Noise Reduction in Speech Processing; Springer: Berlin/Heidelberg, Germany, 2009; pp. 1–4. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Segal, E.; Taskar, B.; Gasch, A.; Friedman, N.; Koller, D. Rich probabilistic models for gene expression. Bioinformatics 2001, 17, S243–S252. [Google Scholar] [CrossRef]
- Ge, F.; Luo, X.; Huang, X.; Zhang, Y.; He, X.; Liu, M.; Lin, H.; Peng, H.; Li, L.; Zhang, Z.; et al. Genome-wide analysis of transcription factors involved in maize embryonic callus formation. Physiol. Plant. 2016, 158, 452–462. [Google Scholar] [CrossRef]
- Gaj, M.D. Factors influencing somatic embryogenesis induction and plant regeneration with particular reference to Arabidopsis thaliana (L.) Heynh. Plant Growth Regul. 2004, 43, 27–47. [Google Scholar] [CrossRef]
- Jin, F.; Hu, L.; Yuan, D.; Xu, J.; Gao, W.; He, L.; Yang, X.; Zhang, X. Comparative transcriptome analysis between somatic embryos (SE s) and zygotic embryos in cotton: Evidence for stress response functions in SE development. Plant Biotechnol. J. 2014, 12, 161–173. [Google Scholar] [CrossRef]
- Thomas, C.; Jiménez, V.M. Mode of action of plant hormones and plant growth regulators during induction of somatic embryogenesis: Molecular aspects. In Somatic Embryogenesis; Springer: Berlin/Heidelberg, Germany, 2005; pp. 157–175. [Google Scholar]
- Yang, X.; Zhang, X.; Yuan, D.; Jin, F.; Zhang, Y.; Xu, J. Transcript profiling reveals complex auxin signalling pathway and transcription regulation involved in dedifferentiation and redifferentiation during somatic embryogenesis in cotton. BMC Plant Biol. 2012, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.E.; Lynch, T.; Peeters, J.; Snowden, C.; Finkelstein, R. A small plant-specific protein family of ABI five binding proteins (AFPs) regulates stress response in germinating Arabidopsis seeds and seedlings. Plant Mol. Biol. 2008, 67, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Nolan, T.M.; Vukašinović, N.; Liu, D.; Russinova, E.; Yin, Y. Brassinosteroids: Multidimensional regulators of plant growth, development, and stress responses. Plant Cell 2020, 32, 295–318. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.B.; Foley, R.C.; Oñate-Sánchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.S.; Dadalto, S.P.; Gonçalves, A.B.; De Souza, G.B.; Barros, V.A.; Fietto, L.G. Plant bZIP transcription factors responsive to pathogens: A review. Int. J. Mol. Sci. 2013, 14, 7815–7828. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.K.; Braam, J.; Fry, S.C.; Nishitani, K. The XTH family of enzymes involved in xyloglucan endotransglucosylation and endohydrolysis: Current perspectives and a new unifying nomenclature. Plant Cell Physiol. 2002, 43, 1421–1435. [Google Scholar] [CrossRef] [PubMed]
- Salaün, C.; Lepiniec, L.; Dubreucq, B. Genetic and molecular control of somatic embryogenesis. Plants 2021, 10, 1467. [Google Scholar] [CrossRef] [PubMed]
- Gulzar, B.; Mujib, A.; Malik, M.Q.; Sayeed, R.; Mamgain, J.; Ejaz, B. Genes, proteins and other networks regulating somatic embryogenesis in plants. J. Genet. Eng. Biotechnol. 2020, 18, 31. [Google Scholar] [CrossRef]
- Hussain, A.; Qarshi, I.A.; Nazir, H.; Ullah, I. Plant tissue culture: Current status and opportunities. In Recent Advances in Plant in Vitro Culture; IntechOpen: London, UK, 2012; Volume 6, pp. 1–28. [Google Scholar]
- Chen, C.J.; Liu, Q.; Zhang, Y.C.; Qu, L.H.; Chen, Y.Q.; Gautheret, D. Genome-wide discovery and analysis of microRNAs and other small RNAs from rice embryogenic callus. RNA Biol. 2011, 8, 538–547. [Google Scholar] [CrossRef]
- Von Aderkas, P.; Bonga, J.M. Influencing micropropagation and somatic embryogenesis in mature trees by manipulation of phase change, stress and culture environment. Tree Physiol. 2000, 20, 921–928. [Google Scholar] [CrossRef]
- Su, Y.H.; Liu, Y.B.; Bai, B.; Zhang, X.S. Establishment of embryonic shoot–root axis is involved in auxin and cytokinin response during Arabidopsis somatic embryogenesis. Front. Plant Sci. 2015, 5, 792. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Song, J.; Zeng, Q.; Ma, Y.; Fang, H.; Yang, L.; Deng, B.; Liu, J.; Fang, J.; Zuo, L.; et al. Auxin and cytokinin mediated regulation involved in vitro organogenesis of papaya. J. Plant Physiol. 2021, 260, 153405. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, D.; Ouyang, K.; Chen, X. High frequency plant regeneration from leaf culture of Neolamarckia cadamba. Plant Biotechnol. 2019, 36, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Leisner, C.P.; Ming, R.; Ainsworth, E.A. Distinct transcriptional profiles of ozone stress in soybean (Glycine max) flowers and pods. BMC Plant Biol. 2014, 14, 335. [Google Scholar]
- He, W.; Zhu, M.r.; Shan, H.y.; Jiang, Y.l.; An, X.l.; Wan, X.Y. Development Regulatory Factors Promoting Efficient Plant Genetic Transformation and Their Application in Maize. China Biotechnol. 2022, 42, 85–98. [Google Scholar]
- Boutilier, K.; Offringa, R.; Sharma, V.K.; Kieft, H.; Ouellet, T.; Zhang, L.; Hattori, J.; Liu, C.M.; Van Lammeren, A.A.; Miki, B.L.; et al. Ectopic expression of BABY BOOM triggers a conversion from vegetative to embryonic growth. Plant Cell 2002, 14, 1737–1749. [Google Scholar] [CrossRef]
- Cheng, S.; Huang, Y.; Zhu, N.; Zhao, Y. The rice WUSCHEL-related homeobox genes are involved in reproductive organ development, hormone signaling and abiotic stress response. Gene 2014, 549, 266–274. [Google Scholar] [CrossRef]
- Horstman, A.; Bemer, M.; Boutilier, K. A transcriptional view on somatic embryogenesis. Regeneration 2017, 4, 201–216. [Google Scholar] [CrossRef]
- Zhao, S.; Jiang, Q.T.; Ma, J.; Zhang, X.W.; Zhao, Q.Z.; Wang, X.Y.; Wang, C.S.; Cao, X.; Lu, Z.X.; Zheng, Y.L.; et al. Characterization and expression analysis of WOX5 genes from wheat and its relatives. Gene 2014, 537, 63–69. [Google Scholar] [CrossRef]
- Debernardi, J.M.; Tricoli, D.M.; Ercoli, M.F.; Hayta, S.; Ronald, P.; Palatnik, J.F.; Dubcovsky, J. A GRF–GIF chimeric protein improves the regeneration efficiency of transgenic plants. Nat. Biotechnol. 2020, 38, 1274–1279. [Google Scholar] [CrossRef]
- Luo, G.; Palmgren, M. GRF-GIF chimeras boost plant regeneration. Trends Plant Sci. 2021, 26, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Dong, H.; Xue, Y.; Su, S.; Wu, Y.; Li, S.; Liu, H.; Li, H.; Han, J.; Shan, X.; et al. Transcriptomic analysis reveals somatic embryogenesis-associated signaling pathways and gene expression regulation in maize (Zea mays L.). Plant Mol. Biol. 2020, 104, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, S.; Zhou, H.; Yuan, Z.; Zhou, T.; Zhang, Y.; Xiang, S.; Yang, F.; Shen, X.; Zhang, D. Transcriptome sequencing analysis of sorghum callus with various regeneration capacities. Planta 2021, 254, 33. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.D.; Guzzo, F.; Toonen, M.A.; Vries, S.C.d. A leucine-rich repeat containing receptor-like kinase marks somatic plant cells competent to form embryos. Development 1997, 124, 2049–2062. [Google Scholar] [CrossRef]
- Ren, H.; Gray, W.M. SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol. Plant 2015, 8, 1153–1164. [Google Scholar] [CrossRef]
- Zanin, F.C.; Freitas, N.C.; Pinto, R.T.; Máximo, W.P.F.; Diniz, L.E.C.; Paiva, L.V. The SAUR gene family in coffee: Genome-wide identification and gene expression analysis during somatic embryogenesis. Mol. Biol. Rep. 2022, 49, 1973–1984. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, K.K.; Tripathi, A.K.; Pareek, A.; Sopory, S.K.; Singla-Pareek, S.L. An improved protocol for efficient transformation and regeneration of diverse indica rice cultivars. Plant Methods 2011, 7, 49. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Q20(%) | Q30(%) | GC(%) |
|---|---|---|---|---|---|
| EC1-1 | 46,199,656 | 45,868,452 | 97.97 | 93.91 | 45.47 |
| EC1-2 | 47,204,670 | 46,872,524 | 98.01 | 94.03 | 45.73 |
| EC1-3 | 45,304,084 | 44,904,798 | 97.98 | 93.89 | 45.89 |
| EC2-1 | 56,352,532 | 55,913,282 | 97.89 | 93.76 | 46.17 |
| EC2-2 | 51,604,738 | 51,177,090 | 97.99 | 94.04 | 46.41 |
| EC2-3 | 47,956,026 | 47,574,058 | 97.85 | 93.60 | 46.05 |
| EC3-1 | 50,727,810 | 50,209,174 | 97.74 | 93.45 | 46.44 |
| EC3-2 | 49,086,688 | 48,718,580 | 97.90 | 93.77 | 46.36 |
| EC3-3 | 42,768,296 | 42,412,912 | 97.80 | 93.53 | 46.06 |
| NEC-1 | 42,526,446 | 42,191,988 | 97.97 | 93.89 | 45.89 |
| NEC-2 | 48,282,194 | 47,874,354 | 97.99 | 94.02 | 45.98 |
| NEC-3 | 45,549,362 | 45,213,084 | 97.92 | 93.74 | 46.00 |
| Total | 714,945,156 | 709,247,312 |
| Type | Unigene | Transcript |
|---|---|---|
| Total number | 131,109 | 195,250 |
| Total base | 113,852,321 | 203,207,453 |
| Largest length (bp) | 14,070 | 14,070 |
| Smallest length (bp) | 201 | 201 |
| Average length (bp) | 868.38 | 1040.76 |
| N50 length (bp) | 1382 | 1710 |
| E90N50 length (bp) | 2683 | 2236 |
| Fragment mapped percent (%) | 58.63 | 83.452 |
| GC percent (%) | 39.27 | 39.97 |
| TransRate score | 0.2618 | 0.37417 |
| BUSCO score | C: 76.9% [S: 72.1%;D: 4.8%] | C: 94.2% [S: 49.7%;D: 44.5%] |
| Data Base | All Unigene Number | Percent (%) |
|---|---|---|
| GO | 42,019 | 32.05 |
| KEGG | 16,027 | 12.22 |
| eggNOG | 41,035 | 31.30 |
| Nr | 51,909 | 39.59 |
| Swiss-Prot | 30,407 | 23.19 |
| Pfam | 27,176 | 20.73 |
| Total_anno | 52,420 | 39.98 |
| Total | 131,109 |
| Sample | Sample | Sample | Pearson Correlation Coefficient (r2) |
|---|---|---|---|
| EC1 | 1 | 2 | 0.969 |
| EC1 | 1 | 3 | 0.976 |
| EC1 | 2 | 3 | 0.971 |
| EC2 | 1 | 2 | 0.991 |
| EC2 | 1 | 3 | 0.970 |
| EC2 | 2 | 3 | 0.966 |
| EC3 | 1 | 2 | 0.996 |
| EC3 | 1 | 3 | 0.986 |
| EC3 | 2 | 3 | 0.992 |
| NEC | 1 | 2 | 0.976 |
| NEC | 1 | 3 | 0.985 |
| NEC | 2 | 3 | 0.990 |
| Family | Number | Proportion | EC1 vs. NEC | EC2 vs. NEC | EC3 vs. NEC | ||||
|---|---|---|---|---|---|---|---|---|---|
| Up | Down | Up | Down | No Change | Up | Down | |||
| MYB superfamily | 64 | 13.73% | 41 | 23 | 36 | 28 | 40 | 24 | |
| AP2/ERF | 56 | 12.02% | 28 | 28 | 29 | 26 | 1 | 30 | 26 |
| bHLH | 48 | 10.30% | 39 | 9 | 37 | 11 | 38 | 10 | |
| NAC | 39 | 8.37% | 10 | 29 | 13 | 25 | 1 | 13 | 26 |
| C2C2 | 35 | 7.51% | 18 | 17 | 8 | 27 | 16 | 19 | |
| WRKY | 29 | 6.22% | 7 | 22 | 7 | 22 | 5 | 24 | |
| LOB | 24 | 5.15% | 13 | 11 | 15 | 9 | 18 | 6 | |
| LBD | 23 | 4.94% | 17 | 6 | 17 | 6 | 18 | 5 | |
| GRAS | 18 | 3.86% | 12 | 6 | 6 | 11 | 1 | 12 | 6 |
| bZIP | 15 | 3.22% | 8 | 7 | 8 | 7 | 9 | 6 | |
| TCP | 14 | 3.00% | 11 | 3 | 9 | 5 | 10 | 4 | |
| B3 superfamily | 13 | 2.79% | 10 | 3 | 7 | 6 | 11 | 2 | |
| HSF | 13 | 2.79% | 5 | 8 | 3 | 9 | 1 | 4 | 9 |
| MADS | 12 | 2.58% | 9 | 3 | 8 | 3 | 1 | 10 | 2 |
| Nin-like | 9 | 1.93% | 9 | 9 | 9 | ||||
| ZF-HD | 8 | 1.72% | 8 | 5 | 2 | 1 | 7 | 1 | |
| GRF | 7 | 1.50% | 5 | 2 | 5 | 2 | 5 | 2 | |
| C2H2 | 6 | 1.29% | 3 | 3 | 5 | 1 | 4 | 2 | |
| NF-Y | 6 | 1.29% | 6 | 6 | 6 | ||||
| FAR1 | 5 | 1.07% | 2 | 3 | 2 | 3 | 2 | 3 | |
| C3H | 5 | 1.07% | 3 | 2 | 3 | 2 | 3 | 2 | |
| SBP | 4 | 0.86% | 4 | 2 | 2 | 4 | |||
| GeBP | 3 | 0.64% | 2 | 1 | 2 | 1 | 2 | 1 | |
| CPP | 3 | 0.64% | 1 | 2 | 1 | 2 | 1 | 2 | |
| SRS | 3 | 0.64% | 3 | 3 | 3 | ||||
| BES1 | 1 | 0.21% | 1 | 1 | 1 | ||||
| S1Fa-like | 1 | 0.21% | 1 | 1 | 1 | ||||
| Whirly | 1 | 0.21% | 1 | 1 | 1 | ||||
| E2F/DP | 1 | 0.21% | 1 | 1 | 1 | ||||
| Sample | CAC | UBQ |
|---|---|---|
| EC1-1 | 24.63 | 20.02 |
| EC1-2 | 23.93 | 19.63 |
| EC1-3 | 24.44 | 18.64 |
| EC3-1 | 23.07 | 18.20 |
| EC3-2 | 24.18 | 19.14 |
| EC3-3 | 25.26 | 19.40 |
| NEC-1 | 24.76 | 19.95 |
| NEC-2 | 24.56 | 19.30 |
| NEC-3 | 25.04 | 19.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linghu, G.; Yu, Z.; Li, M.; Wang, A.; Kang, Y. Comparative Transcriptome Analysis between Embryogenic and Non-Embryogenic Callus of Davidia involucrata. Forests 2023, 14, 1256. https://doi.org/10.3390/f14061256
Linghu G, Yu Z, Li M, Wang A, Kang Y. Comparative Transcriptome Analysis between Embryogenic and Non-Embryogenic Callus of Davidia involucrata. Forests. 2023; 14(6):1256. https://doi.org/10.3390/f14061256
Chicago/Turabian StyleLinghu, Gaoman, Zhaoyou Yu, Meng Li, Anqi Wang, and Yongxiang Kang. 2023. "Comparative Transcriptome Analysis between Embryogenic and Non-Embryogenic Callus of Davidia involucrata" Forests 14, no. 6: 1256. https://doi.org/10.3390/f14061256
APA StyleLinghu, G., Yu, Z., Li, M., Wang, A., & Kang, Y. (2023). Comparative Transcriptome Analysis between Embryogenic and Non-Embryogenic Callus of Davidia involucrata. Forests, 14(6), 1256. https://doi.org/10.3390/f14061256