
Genetic Diversity and Population Structure of Bursaphelenchus xylophilus in Central China Based on SNP Markers
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Purification of Nematodes
2.2. Genome Resequencing
2.3. Identification and Filtration of Mutation Sites
2.4. Genetic Differentiation Analysis
3. Results
3.1. Sample Collection
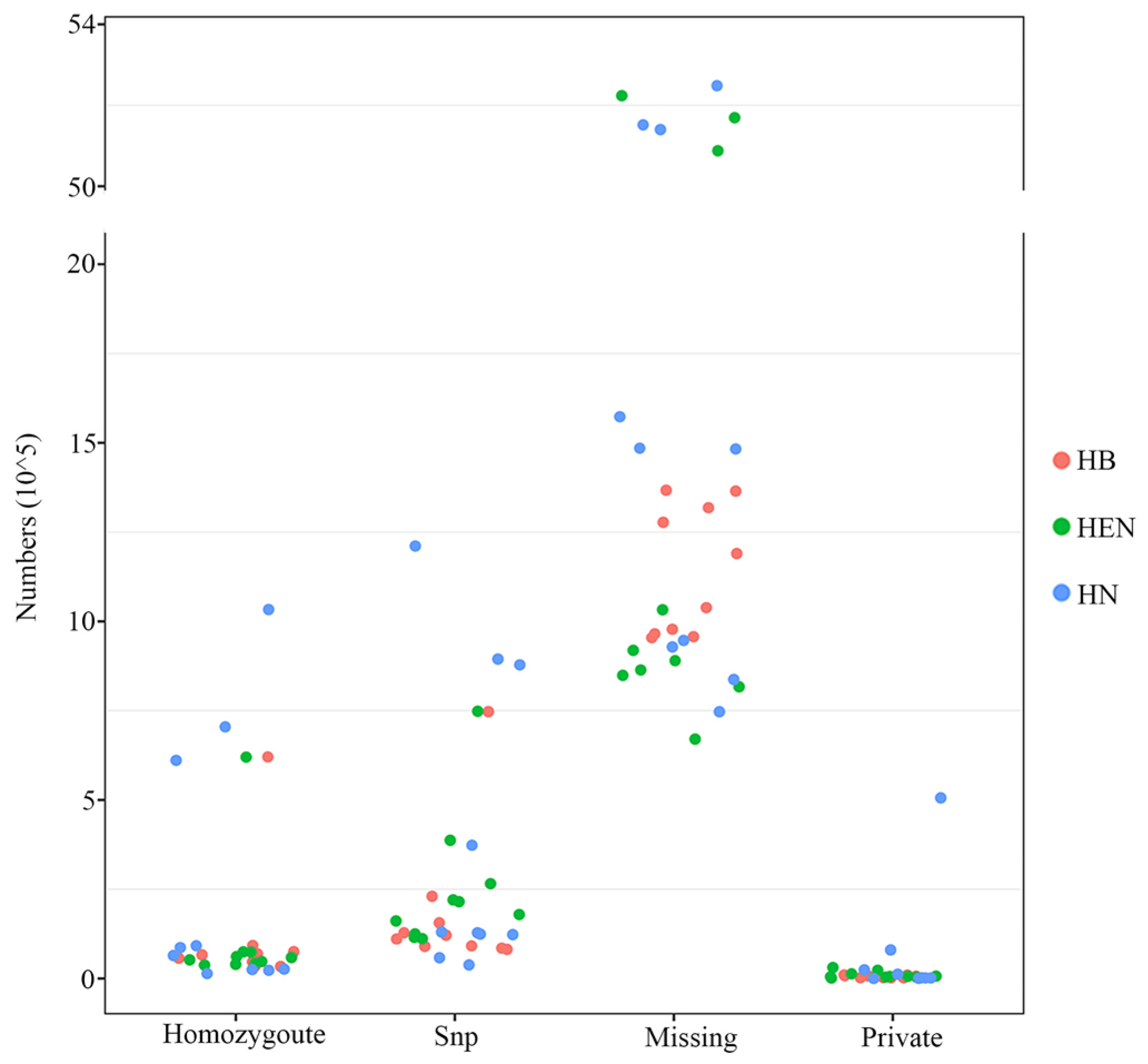
3.2. Statistics of SNP Loci
3.3. Statistics of SNP Genotypes
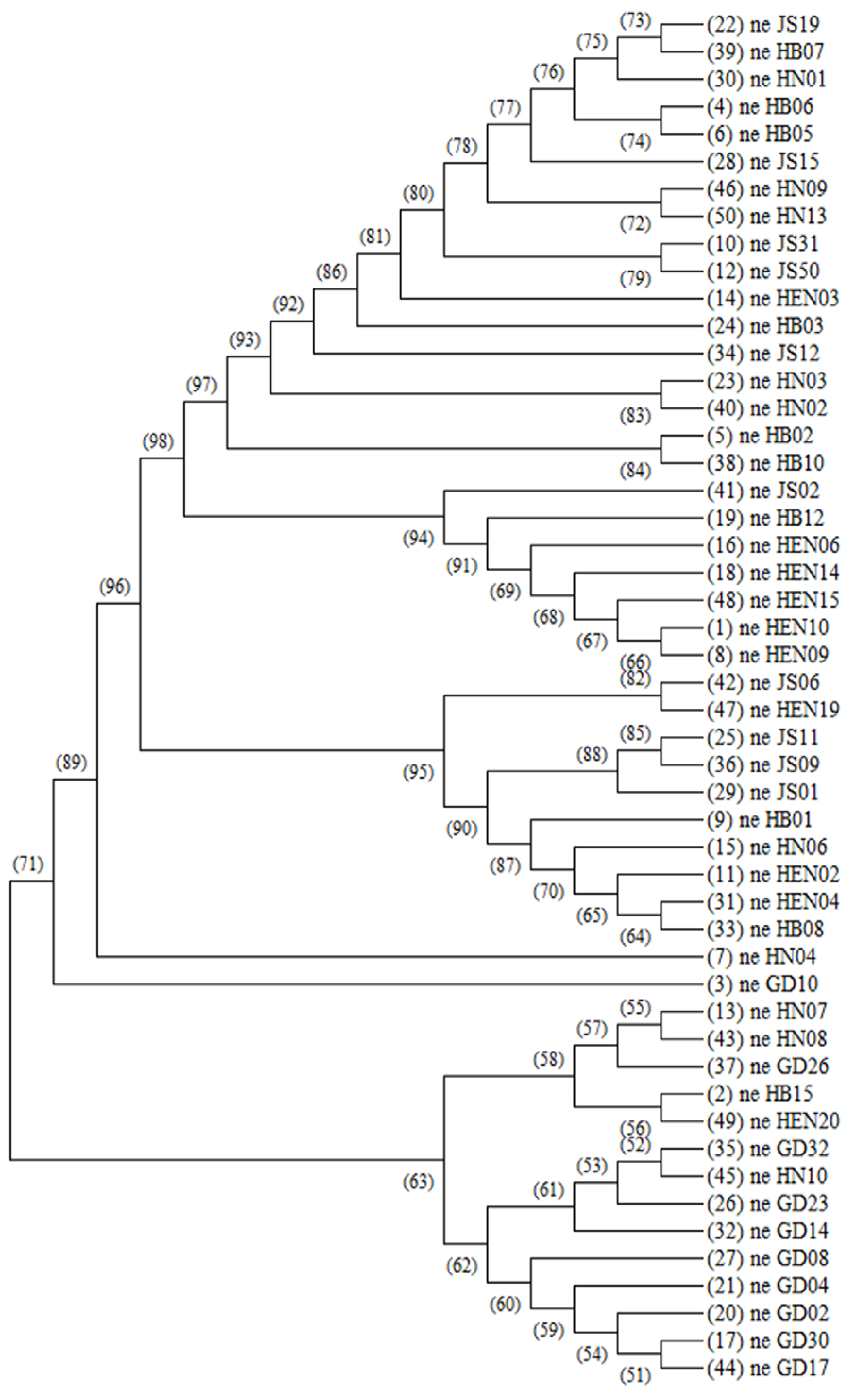
3.4. Analysis of Genetic Differentiation
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, L.X.; Ji, J.; Qiu, X.W.; Zhang L., P. Prevalence and control measures of pine wood nematode disease in the world. China For. Sci. Technol. 2014, 28, 8–13. [Google Scholar]
- Ye, J.R. Epidemic status of pine wilt disease in china and its prevention and control techniques and counter measures. Sci. Silvae Sin. 2019, 55, 1–10. [Google Scholar]
- Ni, A.S.; Yang, D.; Cheng, H.; Ye, J. Preliminary Study on Early Diagnosis and Rehabilitation Treatment of Pine Wood Nematode Disease Based on Partial Symptoms. Forests 2023, 14, 657. [Google Scholar] [CrossRef]
- Wen, T.Y.; Zhang, Y.; Wu, X.Q.; Ye, J.-R.; Qiu, Y.-J.; Rui, L. Studies on the Requirement of Transthyretin Protein (BxTTR-52) for the Suppression of Host Innate Immunity in Bursaphelenchus xylophilus. Int. J. Mol. Sci. 2022, 23, 15058. [Google Scholar] [CrossRef]
- Wang, L.C.; Li, M.; Sheng, R.C.; Chen, F.M. Enzyme-Mediated Amplification (EMA) for Detection of the Pinewood Nematode Bursaphelenchus xylophilus. Forests 2022, 13, 1419. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Lundquist, L.L. Introduction: Population biology, evolution, and control of invasive species. Conserv. Biol. 2003, 17, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Simberloff, D.; Martin, J.; Genovesi, P.; Maris, V.; Wardle, D.A.; Aronson, J.; Courchamp, F.; Galil, B.; García-Berthou, E.; Pascal, M.; et al. Impacts of biological invasions: What’s what and the way forward. Trends Ecol. Evol. 2013, 28, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Estoup, A.; Guillemaud, T. Reconstructing routes of invasion using genetic data: Why, how and so what? Mol. Ecol. 2010, 19, 4113–4130. [Google Scholar] [CrossRef]
- Papura, D.; Burban, C.; Maarten, V.H.; Giresse, X.; Nusillard, B.; Guillemaud, T.; Kerdelhue, C. Microsatellite and mitochondrial data provide evidence for a single major introduction for the Neartic leafhopper Scaphoideus titanus in Europe. PLoS ONE 2012, 7, e36882. [Google Scholar] [CrossRef] [Green Version]
- Boucher, A.C.; Mimee, B.; Montarry, J.; Bardou-Valette, S.; Bélair, G.; Moffett, P.; Grenier, E. Genetic diversity of the golden potato cyst nematode Globodera rostochiensis and determination of the origin of populations in Quebec, Canada. Mol. Phylogenetics Evol. 2013, 69, 75–82. [Google Scholar] [CrossRef]
- Van-Wilgen, B.W.; Moran, V.C.; Hoffmann, J.H. Some perspectives on the risks and benefits of biological control of invasive alien plants in the management of natural ecosystems. Environ. Manag. 2013, 52, 531–540. [Google Scholar] [CrossRef]
- Kikuchi, T.; Cotton, J.A.; Dalzell, J.J.; Hasegawa, K.; Kanzaki, N.; McVeigh, P.; Takanashi, T.; Tsai, I.J.; Assefa, S.A.; Cock, P.J.; et al. Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus. PLoS Pathog. 2011, 7, e1002219. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.S.; Xi, X.T.; Ding, X.L.; Ye, J. Study on the population differentiation of Bursaphelenchus xylophilus in Guangdong province by SNP markers. J. Nanjing For. Univ. 2019, 43, 7. [Google Scholar]
- Xu, J.R.; Wu, X.Q.; Liu, Y. Development of simple sequence repeats base on pine wood nematode (Bursaphelenchus xylophilus) genome sequence. J. Nanjing For. Univ. 2014, 38, 36–42. [Google Scholar]
- Figueiredo, J.; Simões, M.J.; Gomes, P.; Barroso, C.; Pinho, D.; Conceicao, L.; Fonseca, L.; Abrantes, I.; Pinheiro, M.; Egas, C. Assessment of the geographic origins of pine wood nematode isolates via single nucleotide polymorphism in effector genes. PLoS ONE 2013, 8, e83542. [Google Scholar] [CrossRef] [Green Version]
- Vieira, P.; Burgermeister, W.; Mota, M.; Metge, K.; Silva, G. Lack of genetic variation of Bursaphelenchus xylophilus in Portugal revealed by RAPD-PCR analyses. J. Nematol. 2007, 39, 118–126. [Google Scholar]
- Valadas, V.; Laranjo, M.; Barbosa, P.; Espada, M.; Mota, M.; Oliveira, S. The pine wood nematode, Bursaphelenchus xylophilus, in Portugal: Possible introductions and spread routes of a serious biological invasion revealed by molecular methods. Nematology 2012, 14, 899–911. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Xu, Y.H.; Du, J.; Yao, X.W.; Li, Z.D.; Weng, Y.M. Research progress and application of SNP. Sci.-Technol. Innov. Product. 2019, 2, 62–65. [Google Scholar]
- Kwong, Q.B.; Teh, C.K.; Ong, A.L.; Heng, H.Y.; Lee, H.L.; Mohamed, M.; Low, J.Z.B.; Apparow, S.; Chew, F.T.; Mayes, S.; et al. Development and validation of a high-density SNP genotyping array for African oil palm. Mol. Plant 2016, 9, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Ma, D.R.; Tang, L.; Zhao, M.; Zhang, G.; Wang, W.; Song, J.; Li, X.; Liu, Z.; Zhang, W.; et al. Population genomic analysis and de novo assembly reveal the origin of weedy rice as an evolutionary game. Mol. Plant 2019, 12, 632–647. [Google Scholar] [CrossRef] [Green Version]
- Su, T.B.; Wang, W.H.; Li, P.R.; Zhang, B.; Li, P.; Xin, X.; Sun, H.; Yu, Y.; Zhang, D.; Zhao, X.; et al. A genomic variation map provides insights into the genetic basis of spring Chinese cabbage (Brassica rapa ssp. pekinensis) selection. Mol. Plant 2018, 11, 1360–1376. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.T.; Ding, X.L.; Ye, J.R.; Shi, X. Genetic differentiation of Bursaphelenchus xylophilus in east China based on single nucleotide polymorphisms (SNP) markers. J. Nanjing For. Univ. 2022, 46, 21–28. [Google Scholar]
- Ding, X.L.; Ye, J.R.; Lin, S.X.; Wu, X.; Li, D.; Nian, B. Deciphering the molecular variations of pine wood nematode Bursaphelenchus xylophilus with different virulence. PLoS ONE 2016, 11, e0156040. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.Y.; Cheng, X.Y.; Shi, J.; Zhang, Q.W.; Dai, S.M.; Cheng, F.X.; Luo, Y.Q. Formation and expansion mechanism of invasive populations of pine wood nematodes: Progress in “basic research on invasion mechanism and control of dangerous organisms in agriculture and forestry” of national key basic research program. Sci. China 2009, 39, 333–341. [Google Scholar]
- Xie, H. Taxonomy of Plant Nematodes, 2nd ed.; Higher Education Press: Beijing, China, 2005; pp. 305–310. [Google Scholar]
- Chen, F.M.; Ye, J.R.; Wu, X.Q.; Huang, L.; Tang, J. SCAR Marker and Detection Technique of Bursaphelenchus xylophilus. Sci. Silvae Sin. 2012, 48, 88–94. [Google Scholar]
- Cheng, H.R. Occurrence and research progress of pine wood nematode wilt disease. Plant Quar. 1988, 2, 11–17. [Google Scholar]
- Viglierchio, D.R.; Schmitt, R.V. On the methodology of nematode extraction from field samples: Baermann funnel modifications. J. Nematol. 1983, 15, 438–444. [Google Scholar]
- Ding, X.L.; Guo, Y.F.; Ye, J.R.; Wu, X.; Lin, S.; Chen, F.; Zhu, L.; Huang, L.; Song, X.; Zhang, Y.; et al. Population differentiation and epidemic tracking of Bursaphelenchus xylophilus in China based on chromosome-level assembly and whole-genome sequencing data. Pest Manag. Sci. 2022, 78, 1213–1226. [Google Scholar] [CrossRef]
- Huang, Z.H.; Liu, N.F. Advances in Population Genetics. J. Anhui Agric. Sci. 2008, 36, 13490–13491,13499. [Google Scholar]
- Nei, M.; Maruyama, T.; Chakraborty, R. The Bottleneck Effect and Genetic Variability in Populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Li, Y.X.; Zhang, X.Y. Analysis on the trend of invasion and expansion of Bursaphelenchus xylophilus. For. Pest Dis. 2018, 37, 1–4. [Google Scholar]
- Cheng, X.Y.; Cheng, F.X.; Xu, R.M.; Xie, B.Y. Genetic variation in the invasive process of Bursaphelenchus xylophilus (Aphelenchida: Aphelenchoididae) and its possible spread routes in China. Heredity 2008, 100, 356–365. [Google Scholar] [CrossRef]
- Mallez, S.; Castagnone, C.; Espada, M.; Vieira, P.; Eisenback, J.D.; Harrell, M.; Mota, M.; Aikawa, T.; Akiba, M.; Kosaka, H.; et al. Worldwide invasion routes of the pinewood nematode: What can we infer from population genetics analyses? Biol. Invasions 2015, 17, 1199–1213. [Google Scholar] [CrossRef]
- Pan, H.W. Study on the Potential Geographic Distribution of Bursaphelenchus xylophilus in China. Ph.D. Thesis, Chinese Academy of Forestry, Beijing, China, 2009. [Google Scholar]
- Robinet, C.; Roques, A.; Pan, H.Y.; Fang, G.; Ye, J.; Zhang, Y.; Sun, J. Role of human-mediated dispersal in the spread of the pine wood nematode in China. PLoS ONE 2009, 4, e4646. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain No. | Origin | Host | Sampling Date |
|---|---|---|---|
| GD02 | Qingcheng District, Qingyuan City, Guangdong Province | Pinus massoniana | 01/2015 |
| GD04 | Huiyang District, Huizhou City, Guangdong Province | P. massoniana | 01/2015 |
| GD08 | Boluo County, Huizhou City, Guangdong Province | P. massoniana | 01/2015 |
| GD10 | Huangpu District, Guangzhou City, Guangdong Province | P. massoniana | 01/2015 |
| GD14 | Dongguan, Guangdong Province | P. massoniana | 01/2015 |
| GD17 | Tianhe District, Guangzhou City, Guangdong Province | P. massoniana | 01/2015 |
| GD23 | Meijiang District, Meizhou City, Guangdong Province | P. massoniana | 11/2015 |
| GD26 | Fengshun County, Meizhou City, Guangdong Province | P. massoniana | 08/2017 |
| GD30 | Haifeng County, Shanwei City, Guangdong Province | P. massoniana | 08/2017 |
| GD32 | Dongyuan County, Heyuan City, Guangdong Province | P. massoniana | 08/2017 |
| HB01 | Chibi, Xianning City, Hubei Province | P. massoniana | 04/2015 |
| HB02 | Changyang County, Yichang City, Hubei Province | P. massoniana | 04/2015 |
| HB03 | Enshi City, Enshi Prefecture, Hubei Province | P. massoniana | 04/2015 |
| HB05 | Huangpi District, Wuhan City, Hubei Province | P. massoniana | 04/2015 |
| HB06 | Huangpi District, Wuhan City, Hubei Province | P. massoniana | 04/2015 |
| HB07 | Huangpi District, Wuhan City, Hubei Province | P. massoniana | 04/2015 |
| HB08 | Yiling District, Yichang City, Hubei Province | P. massoniana | 11/2015 |
| HB10 | Yidu District, Yichang City, Hubei Province | P. massoniana | 11/2015 |
| HB12 | Zengdu District, Suizhou City, Hubei Province | P. massoniana | 08/2017 |
| HB15 | Luotian County, Huanggang City, Hubei Province | P. massoniana | 08/2017 |
| HEN02 | Xin County, Xinyang City, Henan Province | P. massoniana | 08/2015 |
| HEN03 | Xin County, Xinyang City, Henan Province | P. massoniana | 08/2015 |
| HEN04 | Xin County, Xinyang City, Henan Province | P. massoniana | 08/2015 |
| HEN06 | Xichuan County, Nanyang City, Henan Province | P. massoniana | 10/2017 |
| HEN09 | Xichuan County, Nanyang City, Henan Province | P. massoniana | 11/2017 |
| HEN10 | Xichuan County, Nanyang City, Henan Province | P. massoniana | 11/2017 |
| HEN14 | Xixia County, Nanyang City, Henan Province | Pinus tabuliformis | 01/2018 |
| HEN15 | Xixia County, Nanyang City, Henan Province | P. massoniana | 10/2018 |
| HEN19 | Xin County, Xinyang City, Henan Province | P. massoniana | 10/2018 |
| HEN20 | Xin County, Xinyang City, Henan Province | P. massoniana | 10/2018 |
| HN01 | Cili County, Zhangjiajie City, Hunan Province | P. massoniana | 03/2015 |
| HN02 | Linxiang City, Yueyang City, Hunan Province | P. massoniana | 03/2015 |
| HN03 | Yunxi District, Yueyang City, Hunan Province | P. massoniana | 03/2015 |
| HN04 | Hengnan County, Hengyang City, Hunan Province | P. massoniana | 03/2015 |
| HN06 | Cili County, Zhangjiajie City, Hunan Province | P. massoniana | 08/2015 |
| HN07 | Taoyuan County, Changde City, Hunan Province | P. massoniana | 08/2016 |
| HN08 | Taoyuan County, Changde City, Hunan Province | P. massoniana | 08/2016 |
| HN09 | Lingling District, Yongzhou City, Hunan Province | P. massoniana | 03/2019 |
| HN10 | Shaoyang County, Shaoyang City, Hunan Province | P. massoniana | 03/2019 |
| HN13 | Lingling District, Yongzhou City, Hunan Province | P. massoniana | 03/2019 |
| JS01 | Liuhe District, Nanjing City, Jiangsu Province | P. massoniana | 12/2014 |
| JS02 | Runzhou District, Zhenjiang City, Jiangsu Province | P. massoniana | 12/2014 |
| JS06 | Binhu District, Wuxi City, Jiangsu Province | P. massoniana | 12/2014 |
| JS09 | Xuyi County, Huai‘an City, Jiangsu Province | P. massoniana | 01/2015 |
| JS11 | Haizhou District, Lianyungang City, Jiangsu Province | Pinus densiflora | 01/2015 |
| JS12 | Yizheng, Yangzhou City, Jiangsu Province | P. massoniana | 01/2015 |
| JS15 | Changshu City, Suzhou City, Jiangsu Province | P. massoniana | 02/2015 |
| JS19 | Runzhou District, Zhenjiang City, Jiangsu Province | P. massoniana | 10/2017 |
| JS31 | Lishui District, Nanjing City, Jiangsu Province | P. massoniana | 10/2017 |
| JS50 | Jintan District, Changzhou City, Jiangsu Province | P. massoniana | 10/2017 |
| Strain No. | SNP Count | Homozygous | Missing | Specific SNP Count |
|---|---|---|---|---|
| HB01 | 81,723 | 46,927 | 1,367,096 | 6644 |
| HB02 | 110,859 | 75,239 | 1,189,888 | 6547 |
| HB03 | 128,236 | 34,233 | 957,159 | 2695 |
| HB05 | 90,192 | 45,461 | 1,318,256 | 2291 |
| HB06 | 91,540 | 57,049 | 965,177 | 2290 |
| HB07 | 121,555 | 69,319 | 1,038,514 | 9634 |
| HB08 | 85,121 | 33,889 | 1,277,353 | 2195 |
| HB10 | 230,409 | 66,327 | 954,380 | 7499 |
| HB12 | 156,058 | 92,696 | 978,007 | 8018 |
| HB15 | 747,217 | 620,622 | 1,364,706 | 10,220 |
| HEN02 | 179,367 | 39,985 | 890,120 | 6229 |
| HEN03 | 115,598 | 73,932 | 1,032,402 | 13,361 |
| HEN04 | 161,298 | 59,004 | 863,933 | 6837 |
| HEN06 | 387,158 | 74,788 | 670,407 | 31,052 |
| HEN09 | 265,742 | 52,417 | 848,798 | 23,215 |
| HEN10 | 215,398 | 41,954 | 816,941 | 6011 |
| HEN14 | 111,679 | 61,536 | 918,903 | 4289 |
| HEN15 | 125,089 | 47,713 | 5,169,332 | 1586 |
| HEN19 | 220,318 | 37,791 | 5,087,640 | 5128 |
| HEN20 | 748,317 | 620,041 | 5,223,813 | 3968 |
| HN01 | 123,168 | 86,716 | 946,562 | 1316 |
| HN02 | 38,277 | 14,055 | 1,572,950 | 716 |
| HN03 | 124,574 | 91,984 | 928,412 | 1906 |
| HN04 | 58,334 | 25,275 | 1,484,882 | 2413 |
| HN06 | 373,296 | 26,536 | 1,482,689 | 80,083 |
| HN07 | 894,621 | 704,750 | 747,078 | 24,547 |
| HN08 | 878,428 | 611,007 | 837,408 | 12,192 |
| HN09 | 130,567 | 23,176 | 5,151,805 | 396 |
| HN10 | 1,210,980 | 1,033,119 | 5,248,382 | 505,849 |
| HN13 | 128,256 | 64,496 | 5,140,161 | 1855 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, A.; Ding, X.; Feng, Y.; Chen, T.; Ye, J. Genetic Diversity and Population Structure of Bursaphelenchus xylophilus in Central China Based on SNP Markers. Forests 2023, 14, 1443. https://doi.org/10.3390/f14071443
Yang A, Ding X, Feng Y, Chen T, Ye J. Genetic Diversity and Population Structure of Bursaphelenchus xylophilus in Central China Based on SNP Markers. Forests. 2023; 14(7):1443. https://doi.org/10.3390/f14071443
Chicago/Turabian StyleYang, Aixia, Xiaolei Ding, Yuan Feng, Tingting Chen, and Jianren Ye. 2023. "Genetic Diversity and Population Structure of Bursaphelenchus xylophilus in Central China Based on SNP Markers" Forests 14, no. 7: 1443. https://doi.org/10.3390/f14071443