
The Genome-Wide Profiling of Alternative Splicing in Willow under Salt Stress

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatment
2.2. Physiological Index Test
2.3. RNA Extraction and Library Construction
2.4. RNA-Seq and Data Analysis
2.5. RT–PCR Validation
3. Results
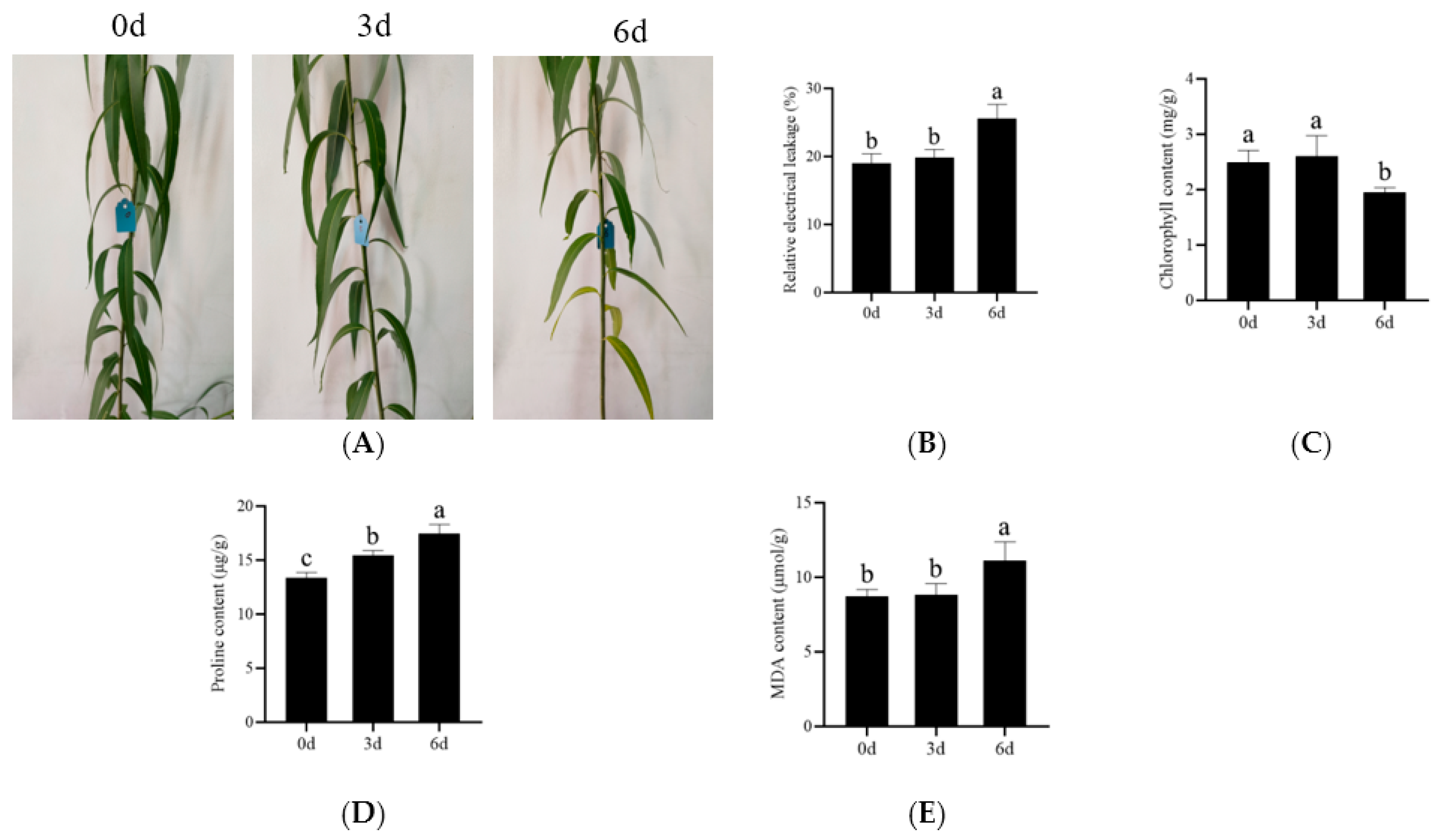
3.1. Physiological Performance of S. matsudana Koidz 9901 under Salt Stress
3.2. Overview of Iso-Seq Data
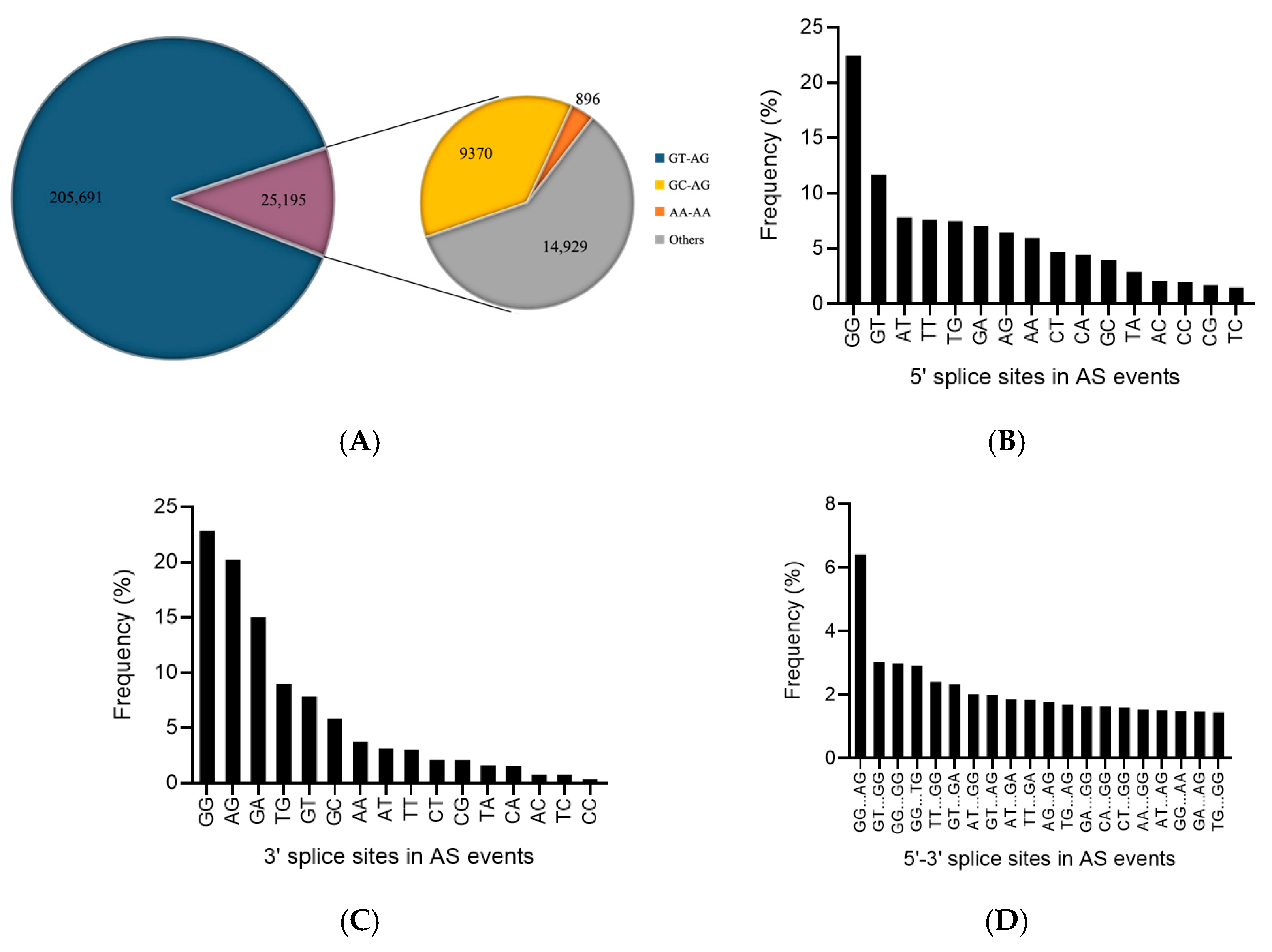
3.3. AS Events in 9901 Willow under Salt Stress
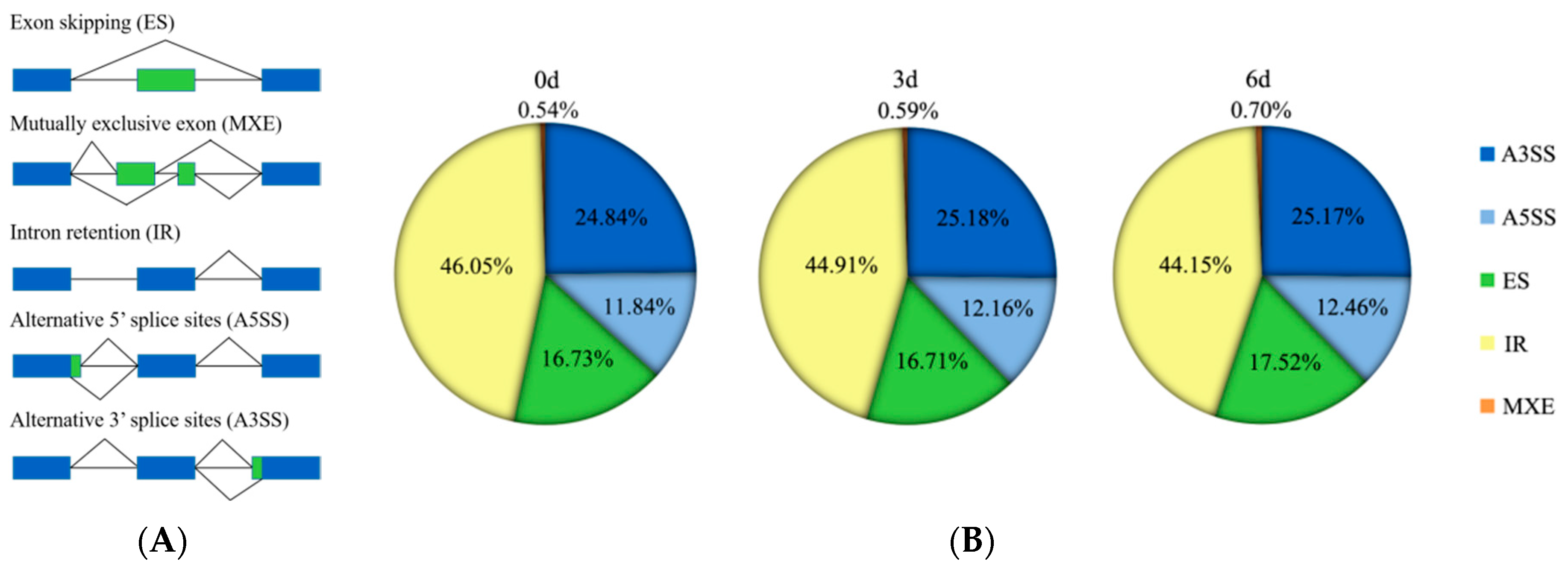
3.4. Alternative Splicing Modes of the 9901 Willow Genes
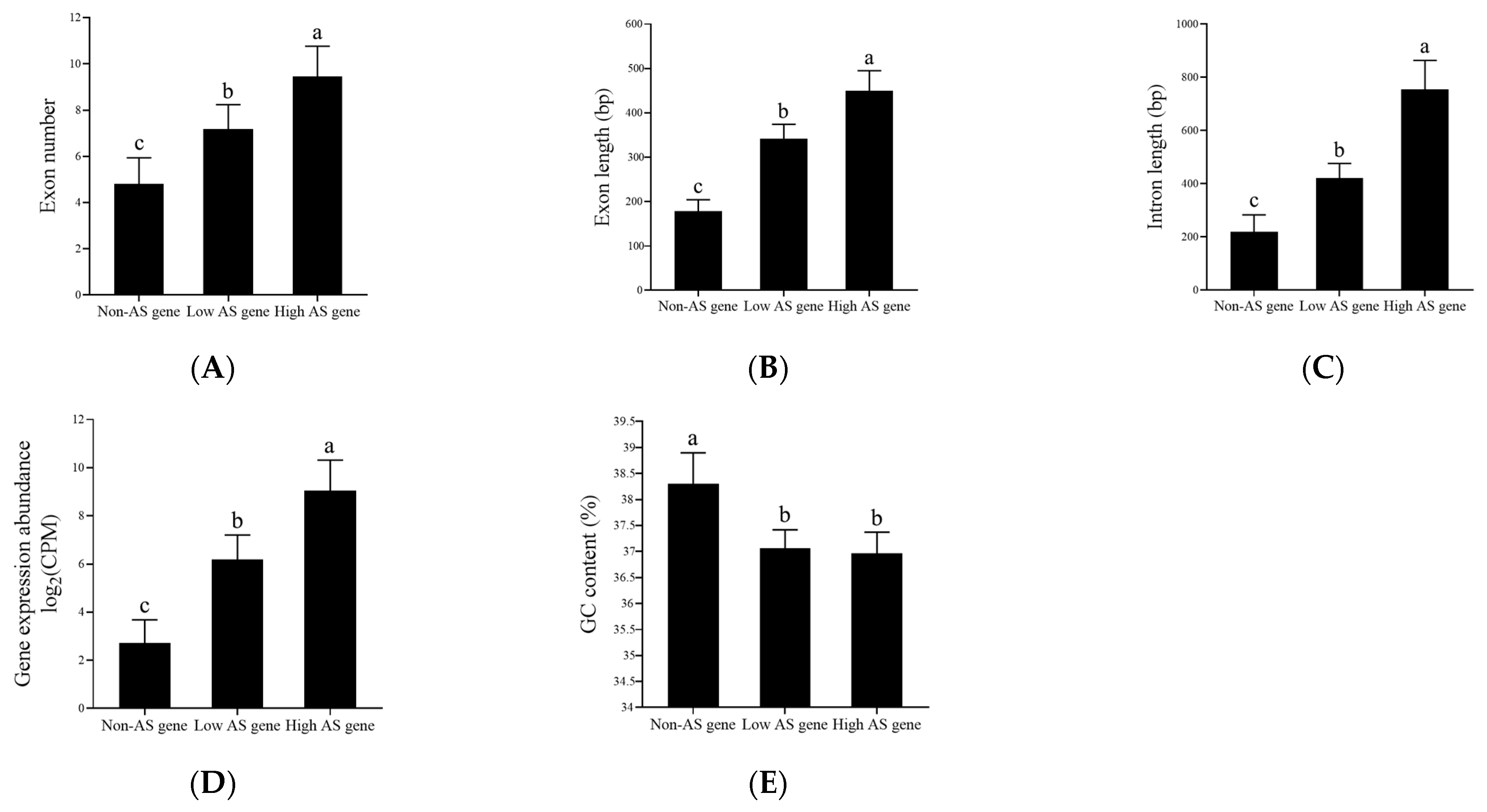
3.5. Characterization of AS Genes
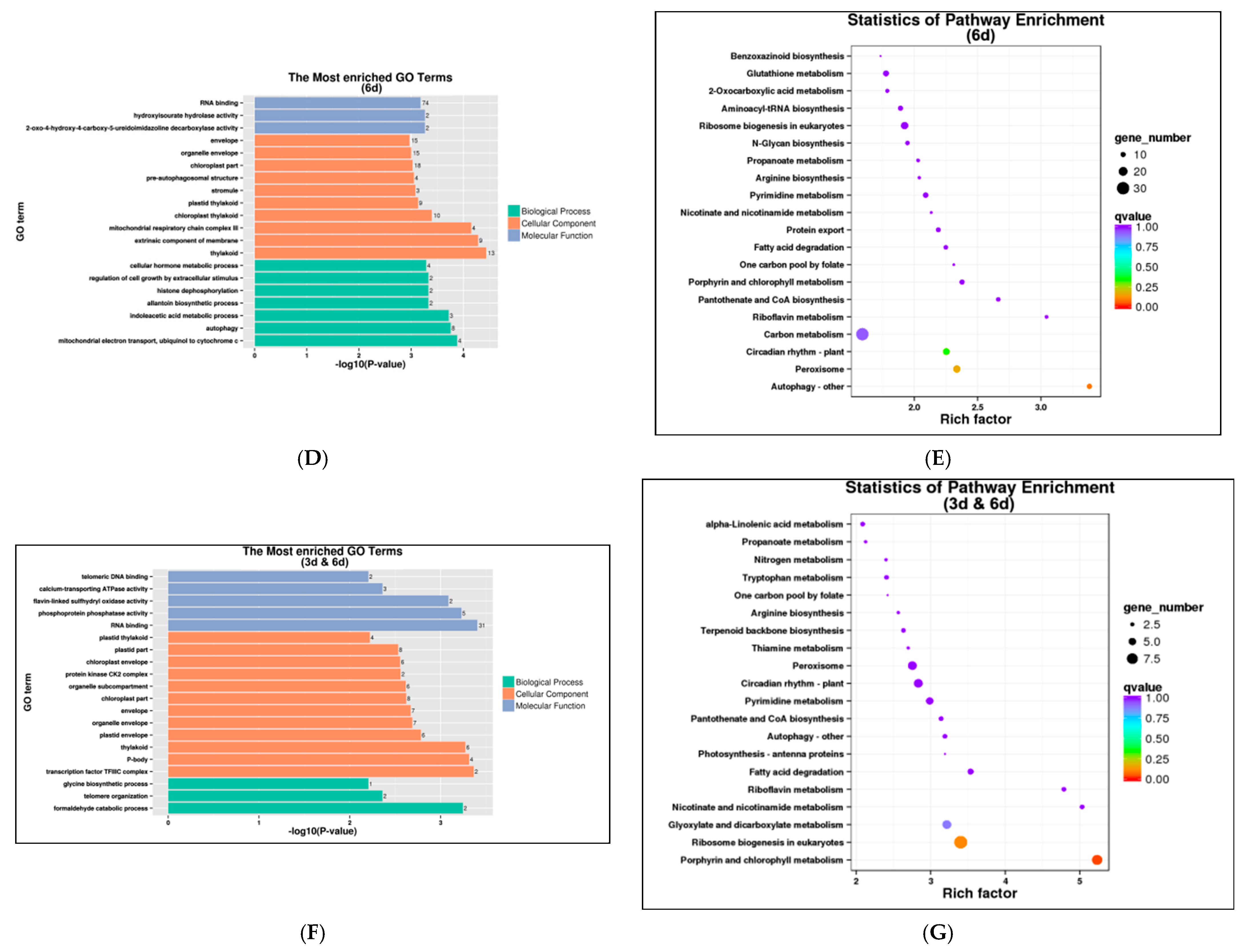
3.6. AS Performance of Willow Genes during Salt Stress
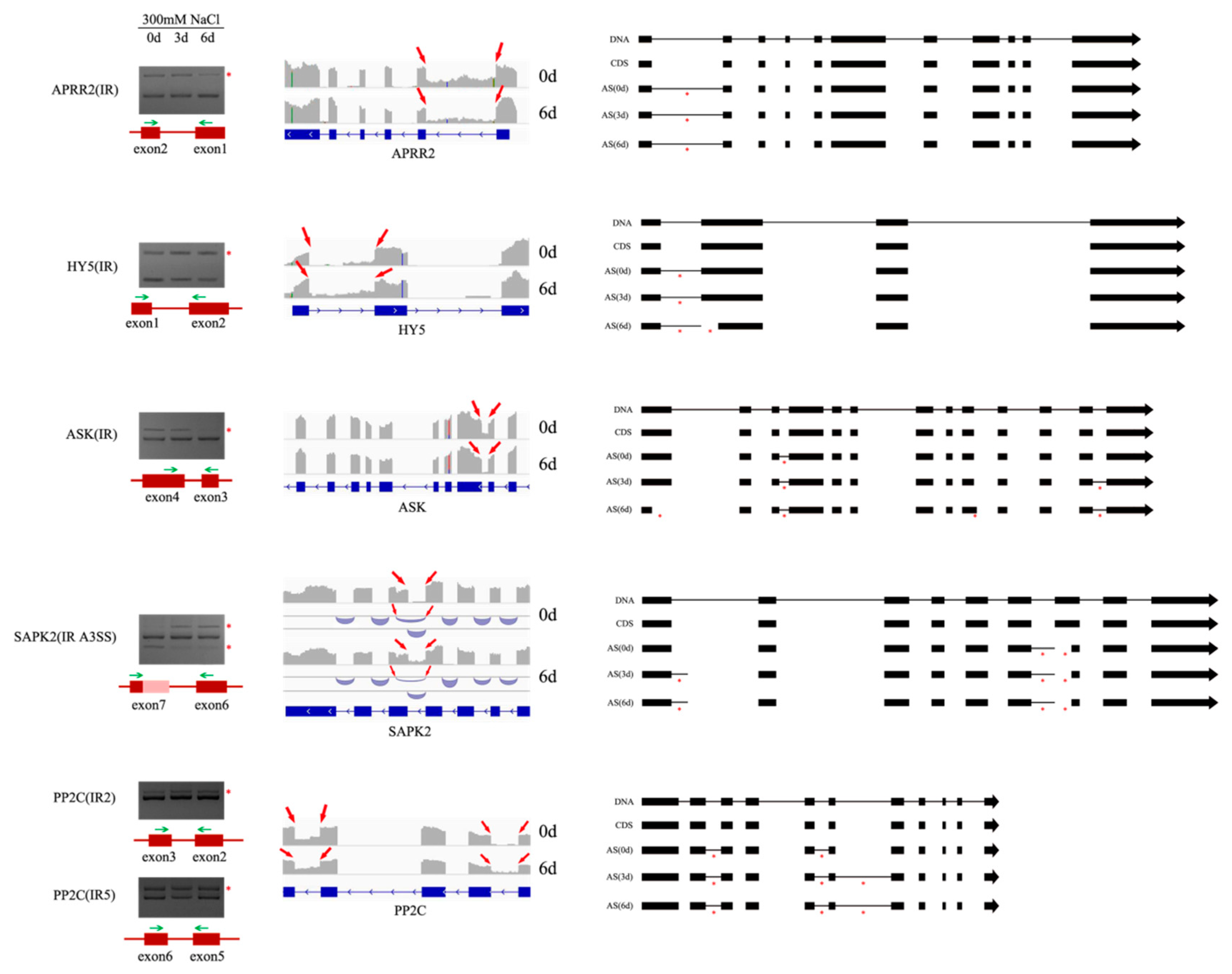
3.7. AS Patterns of Five Genes during Salt Stress
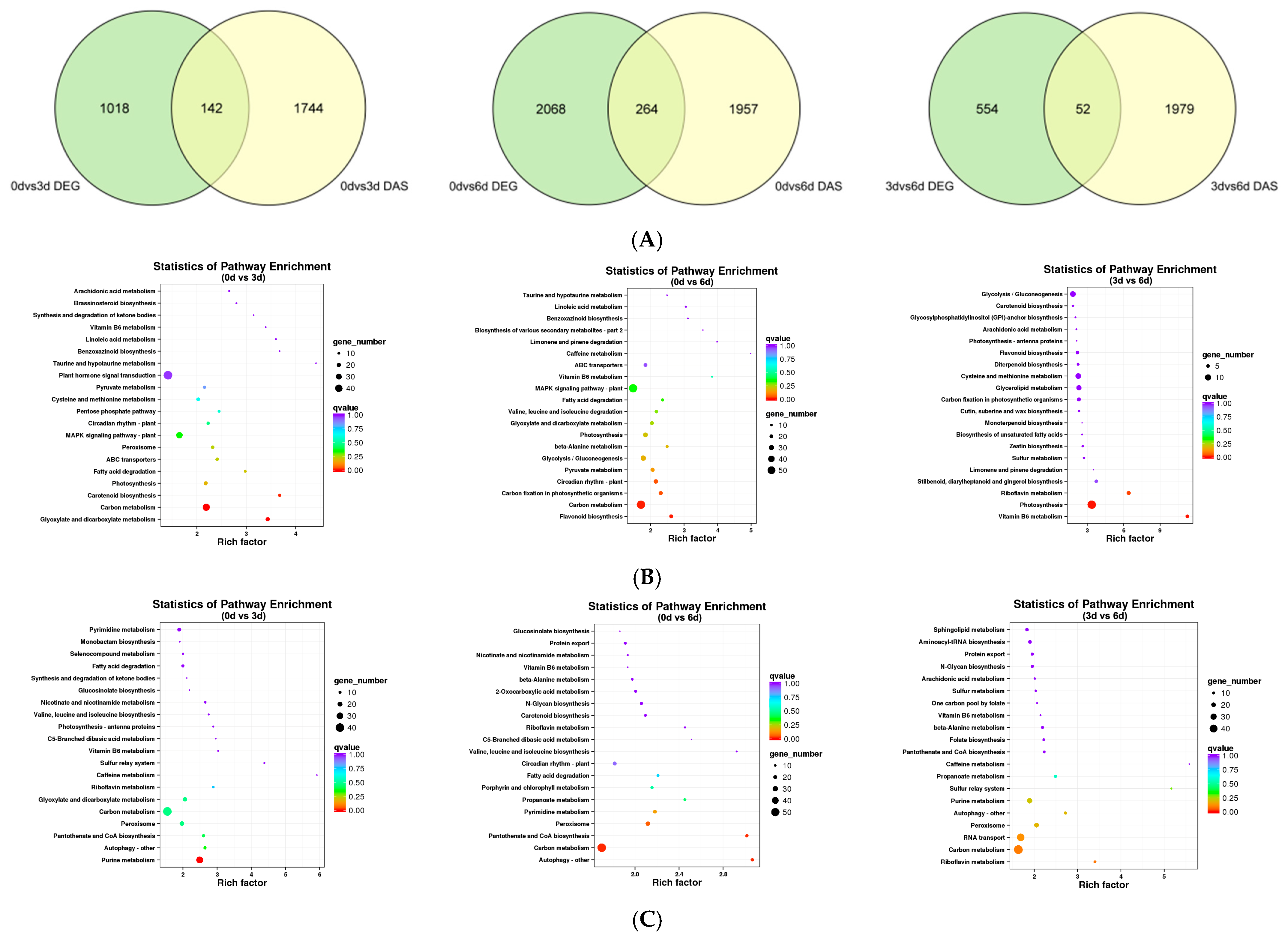
3.8. Evaluation of Differentially Expressed Genes (DEGs) and Differential AS (DAS) Genes against Salt Stress in 9901 Willow
4. Discussion
4.1. Characterization of AS Genes in Willow
4.2. Abiotic Stress Advanced the AS Events as a Mechanism of Plant Decline
4.3. AS Effects on Salt Resistance of Plants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Godoy Herz, M.A.; Kornblihtt, A.R. Alternative splicing and transcription elongation in plants. Front. Plant Sci. 2019, 10, 309. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Halter, G.; Renner, T.; Waters, E.R. Patterns of alternative splicing vary between species during heat stress. AoB Plants 2018, 10, ply013. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Shi, J.; Zhang, Z.; Zhong, B.; Zhu, Z. Plant AFC2 kinase desensitizes thermomorphogenesis through modulation of alternative splicing. Iscience 2022, 25, 104051. [Google Scholar] [CrossRef] [PubMed]
- Dikaya, V.; El Arbi, N.; Rojas-Murcia, N.; Nardeli, S.M.; Goretti, D.; Schmid, M. Insights into the role of alternative splicing in plant temperature response. J. Exp. Bot. 2021, 72, 7384–7403. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhou, Z.; Wang, Z.; Li, W.; Fang, C.; Wu, M.; Ma, Y.; Liu, T.; Kong, L.A.; Peng, D.L.; et al. Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hu, T.; Li, X.; Mu, S.; Cheng, Z.; Ge, W.; Gao, J. Genome-wide analysis of shoot growth-associated alternative splicing in moso bamboo. Mol. Genet. Genom. 2016, 291, 1695–1714. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Kong, Y.; Song, D.; Huang, C.; Li, X.; Li, L. Conservation and functional influence of alternative splicing in wood formation of Populus and Eucalyptus. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, X.; Cheng, Z.; Zeng, C.; Li, W.; Zhang, L.; Peng, M. Large-scale analysis of the cassava transcriptome reveals the impact of cold stress on alternative splicing. J. Exp. Bot. 2020, 71, 422–434. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, D.; Wang, S.; Yang, L. Large-scale detection and application of expressed sequence tag single nucleotide polymorphisms in Nicotiana. Genet. Mol. Res. 2015, 14, 7793–7800. [Google Scholar] [CrossRef]
- Qin, Z.; Wu, J.; Geng, S.; Feng, N.; Chen, F.; Kong, X.; Song, G.; Chen, K.; Li, A.; Mao, L.; et al. Regulation of FT splicing by an endogenous cue in temperate grasses. Nat. Commun. 2017, 8, 14320. [Google Scholar] [CrossRef]
- Wang, H.; Bian, M.; Yang, Z.; Lin, C.; Shi, W. Preliminary functional analysis of the isoforms of OsHsfA2a (Oryza sativa L.) generated by alternative splicing. Plant Mol. Biol. Rep. 2013, 31, 38–46. [Google Scholar] [CrossRef]
- Matsukura, S.; Mizoi, J.; Yoshida, T.; Todaka, D.; Ito, Y.; Maruyama, K.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Comprehensive analysis of rice DREB2-type genes that encode transcription factors involved in the expression of abiotic stress-responsive genes. Mol. Genet. Genom. 2010, 283, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, A.; Punzo, P.; Van Oosten, M.J.; Cirillo, V.; Esposito, S.; Costa, A.; Maggio, A.; Grillo, S.; Batelli, G. Transcriptomic and splicing changes underlying tomato responses to combined water and nutrient stress. Front. Plant Sci. 2022, 13, 974048. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Xie, Y.; Li, J.; Li, Y.; Chen, Q. Research progress of alternative splicing in plants and animals. Genom. Appl. Biol. 2020, 39, 1595–1600. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Wang, R.; Gong, L.; Wang, L.; Xu, J. Genome-wide analysis of alternative splicing events responding to high temperatures in Populus tomentosa Carr. Forests 2023, 14, 1878. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, R.; Wang, Y.; Wang, X.; Wang, L.; Xu, J. The expansin gene SmEXPA13 in Salix matsudana in association with plant salt tolerance. Plant Cell Tissue Organ Cult. 2023, 154, 219–225. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, H.; Yang, R.; Xu, X.; Liu, X.; Xu, J. Over-expression of PttEXPA8 gene showed various resistances to diverse stresses. Int. J. Biol. Macromol. 2019, 130, 50–57. [Google Scholar] [CrossRef]
- Zhang, H.; Shi, Y.; Liu, X.; Wang, R.; Li, J.; Xu, J. Transgenic creeping bentgrass plants expressing a Picea wilsonii dehydrin gene (PicW) demonstrate improved freezing tolerance. Mol. Biol. Rep. 2018, 45, 1627–1635. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, H.; Li, Y.; Chen, Y.; Liu, G.; Ye, M.; Yu, C.; Lian, B.; Zhong, F.; Jiang, Y.; et al. Genome sequencing and phylogenetic analysis of allotetraploid Salix matsudana Koidz. Hortic. Res. 2020, 7, 201. [Google Scholar] [CrossRef]
- Foissac, S.; Sammeth, M. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, W297–W299. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Li, W.; Zhang, F.; Guo, W. RNA-seq analysis reveals alternative splicing under salt stress in cotton, Gossypium davidsonii. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yin, P.; Ruixia, Y.; Liu, X.; Luo, L.; Xu, J. Genome-wide profiling of alternative splicing genes in hybrid poplar (P. alba×P. glandulosa cv.84K) leaves. PLoS ONE 2020, 15, e0241914. [Google Scholar] [CrossRef]
- Hu, H.; Yang, W.; Zheng, Z.; Niu, Z.; Yang, Y.; Wan, D.; Liu, J.; Ma, T. Analysis of alternative splicing and alternative polyadenylation in Populus alba var. pyramidalis by single-molecular long-read sequencing. Front. Genet. 2020, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.B.; Brendel, V. Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. USA 2006, 103, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.Y.; Chen, X.; Song, Y.C.; Lam, L.P.Y.; Tobimatsu, Y.; Gao, B.; Chen, M.X.; Cao, F.L. SWATH-MS-based proteogenomic analysis reveals the involvement of alternative splicing in poplar upon lead stress. Genome Res. 2023, 33, 371–385. [Google Scholar] [CrossRef]
- Cheng, W.; Hong, C.; Zeng, F.; Liu, N.; Gao, H. Sequence variations affect the 5’ splice site selection of plant introns. Plant Physiol. 2023, 193, 1281–1296. [Google Scholar] [CrossRef]
- Thatcher, S.R.; Danilevskaya, O.N.; Meng, X.; Beatty, M.; Zastrow-Hayes, G.; Harris, C.; Van Allen, B.; Habben, J.; Li, B. Genome-wide analysis of alternative splicing during development and drought stress in maize. Plant Physiol. 2016, 170, 586–599. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Q.; Liu, P.; Huang, J.; Zhang, S.; Yang, G.; Wu, C.; Zheng, C.; Yan, K. Dual roles of the serine/arginine-rich splicing factor SR45a in promoting and interacting with nuclear cap-binding complex to modulate the salt-stress response in Arabidopsis. New Phytol. 2021, 230, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Marcella, D.F.A.; Willing, E.M.; Szabo, E.X.; Francisco-Mangilet, A.G.; Droste-Borel, I.; Maček, B.; Schneeberger, K.; Laubinger, S. The U1 snRNP subunit LUC7 modulates plant development and stress responses via regulation of alternative splicing. Plant Cell 2018, 30, 2838–2854. [Google Scholar] [CrossRef]
- Albaqami, M.; Laluk, K.; Reddy, A.S. The Arabidopsis splicing regulator SR45 confers salt tolerance in a splice isoform-dependent manner. Plant Mol. Biol. 2019, 100, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Cui, P.; Wang, Z.; Zhang, S.; Ali, S.; Xiong, L. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genom. 2014, 15, 431. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Lv, X.; Sun, Y.; Fan, Z.; Xiang, G.; Yao, Y. Comprehensive discovery of salt-responsive alternative splicing events based on Iso-Seq and RNA-seq in grapevine roots. Environ. Exp. Bot. 2021, 192, 104645. [Google Scholar] [CrossRef]
- Liu, Z.; Qin, J.; Tian, X.; Xu, S.; Wang, Y.; Li, H.; Wang, X.; Peng, H.; Yao, Y.; Hu, Z.; et al. Global profiling of alternative splicing landscape responsive to drought, heat and their combination in wheat (Triticum aestivum L.). Plant Biotechnol. J. 2018, 16, 714–726. [Google Scholar] [CrossRef]
- Sook Lee, S.; Yong Jung, W.; Ji Park, H.; Lee, A.; Kwon, S.Y.; Kim, H.S.; Sun Cho, H. Genome-wide analysis of alternative splicing in an inbred cabbage (Brassica oleracea L.) line ‘HO’ in response to heat stress. Curr. Genom. 2018, 19, 12–20. [Google Scholar] [CrossRef]
- Lu, F.; Li, W.; Peng, Y.; Cao, Y.; Qu, J.; Sun, F.; Yang, Q.; Lu, Y.; Zhang, X.; Zhng, L.; et al. ZmPP2C26 alternative splicing variants negatively regulate drought tolerance in maize. Front. Plant Sci. 2022, 13, 851531. [Google Scholar] [CrossRef]
- Mitchell, S.F.; Parker, R. Principles and properties of eukaryotic mRNPs. Mol. Cell 2014, 54, 547–558. [Google Scholar] [CrossRef]
- Aneeta Sanan-Mishra, N.; Tuteja, N.; Sopory, S.K. Salinity-and ABA-induced up-regulation and light-mediated modulation of mRNA encoding glycine-rich RNA-binding protein from Sorghum bicolor. Biochem. Biophys. Res. Commun. 2002, 296, 1063–1068. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, D.W.; Wang, Y.C.; Zheng, L.; Yang, C.P. A glycine-rich RNA-binding protein can mediate physiological responses in transgenic plants under salt stress. Mol. Biol. Rep. 2012, 39, 1047–1053. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, X.; Wang, B.; Liu, E.; Chen, N.; Zhang, W.; Liu, H. Overexpression of an Arabidopsis heterogeneous nuclear ribonucleoprotein gene, AtRNP1, affects plant growth and reduces plant tolerance to drought and salt stresses. Biochem. Biophys. Res. Commun. 2016, 472, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, X.; Xu, Q.; Mei, X.; Yuan, H.; Jiabu, D.; Sang, Z.; Nyima, T. Metabolite profiling in two contrasting Tibetan hulless barley cultivars revealed the core salt-responsive metabolome and key salt-tolerance biomarkers. AoB Plants 2019, 11, 021. [Google Scholar] [CrossRef] [PubMed]
- Sukrong, S.; Yun, K.Y.; Stadler, P.; Kumar, C.; Facciuolo, T.; Moffatt, B.A.; Falcone, D.L. Improved growth and stress tolerance in the Arabidopsis oxt1 mutant triggered by altered adenine metabolism. Mol. Plant 2012, 5, 1310–1332. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, Y.; Park, S.Y.; Kim, Y.S.; Wang, S.H.; Yoo, S.C.; Hörtensteiner, S.; Paek, N.C. Arabidopsis STAY-GREEN2 is a negative regulator of chlorophyll degradation during leaf senescence. Mol. Plant 2014, 7, 1288–1302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, Z.; Zhang, X.; Liu, B.; Wang, S.; Wang, Y. Molecular cloning of MhPPOX1 gene from Malus halliana and anti-iron deficiency function identification. Acta Bot. Boreali-Occident. Sin. 2020, 40, 1627–1637. [Google Scholar] [CrossRef]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef]
- Memisoglu, G.; Eapen, V.V.; Yang, Y.; Klionsky, D.J.; Haber, J.E. PP2C phosphatases promote autophagy by dephosphorylation of the Atg1 complex. Proc. Natl. Acad. Sci. USA 2019, 116, 1613–1620. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scheme * | Number of Clean Reads | Number of Full Length Reads | Full-Length Percentage | Number of Mapped Reads | Mapped Rates |
|---|---|---|---|---|---|
| 0-1 | 5,376,795 | 4,976,324 | 92.55% | 4,813,677 | 96.73% |
| 0-2 | 5,651,929 | 5,249,208 | 92.87% | 5,093,243 | 97.03% |
| 0-3 | 6,263,598 | 5,885,644 | 93.97% | 5,504,312 | 93.52% |
| 3-1 | 4,546,552 | 4,209,596 | 92.59% | 4,061,050 | 96.47% |
| 3-2 | 5,471,691 | 5,085,932 | 92.95% | 4,923,362 | 96.80% |
| 3-3 | 6,171,131 | 5,681,920 | 92.07% | 5,172,763 | 91.04% |
| 6-1 | 5,561,594 | 5,144,088 | 92.49% | 5,015,632 | 97.50% |
| 6-2 | 6,071,375 | 5,607,172 | 92.35% | 5,455,723 | 97.30% |
| 6-3 | 5,127,798 | 4,712,937 | 91.91% | 4,582,490 | 97.23% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Gong, L.; Zhang, J.; Wang, L.; Wu, D.; Xu, J. The Genome-Wide Profiling of Alternative Splicing in Willow under Salt Stress. Forests 2024, 15, 30. https://doi.org/10.3390/f15010030
Wang X, Gong L, Zhang J, Wang L, Wu D, Xu J. The Genome-Wide Profiling of Alternative Splicing in Willow under Salt Stress. Forests. 2024; 15(1):30. https://doi.org/10.3390/f15010030
Chicago/Turabian StyleWang, Xue, Longfeng Gong, Junkang Zhang, Lei Wang, Di Wu, and Jichen Xu. 2024. "The Genome-Wide Profiling of Alternative Splicing in Willow under Salt Stress" Forests 15, no. 1: 30. https://doi.org/10.3390/f15010030
APA StyleWang, X., Gong, L., Zhang, J., Wang, L., Wu, D., & Xu, J. (2024). The Genome-Wide Profiling of Alternative Splicing in Willow under Salt Stress. Forests, 15(1), 30. https://doi.org/10.3390/f15010030