
Genome-Wide Identification and Expression Profiling of the Response Regulator (RR) Gene Family in Pecan Reveals Its Possible Association with Callus Formation during Grafting
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Detection of Trans-Zeatin Riboside (tZR) during Grafting
2.2. Genome-Wide Identification of CiRR Members
2.3. Physio-Chemical and Phylogenetic Analyses of the CiRR Gene Family
2.4. Exploring the Structure, Duplication Event, Selective Pressure, and Cis-Element of CiRRs
2.5. Expression Profile Analysis of CiRRs in Different Tissues and Response to Stress and Development
2.6. Investigating the Potential Role of CiRRs during Grafting
3. Results
3.1. Dynamic Changes of tZR during Grafting
3.2. Total Members for the CiRR Family
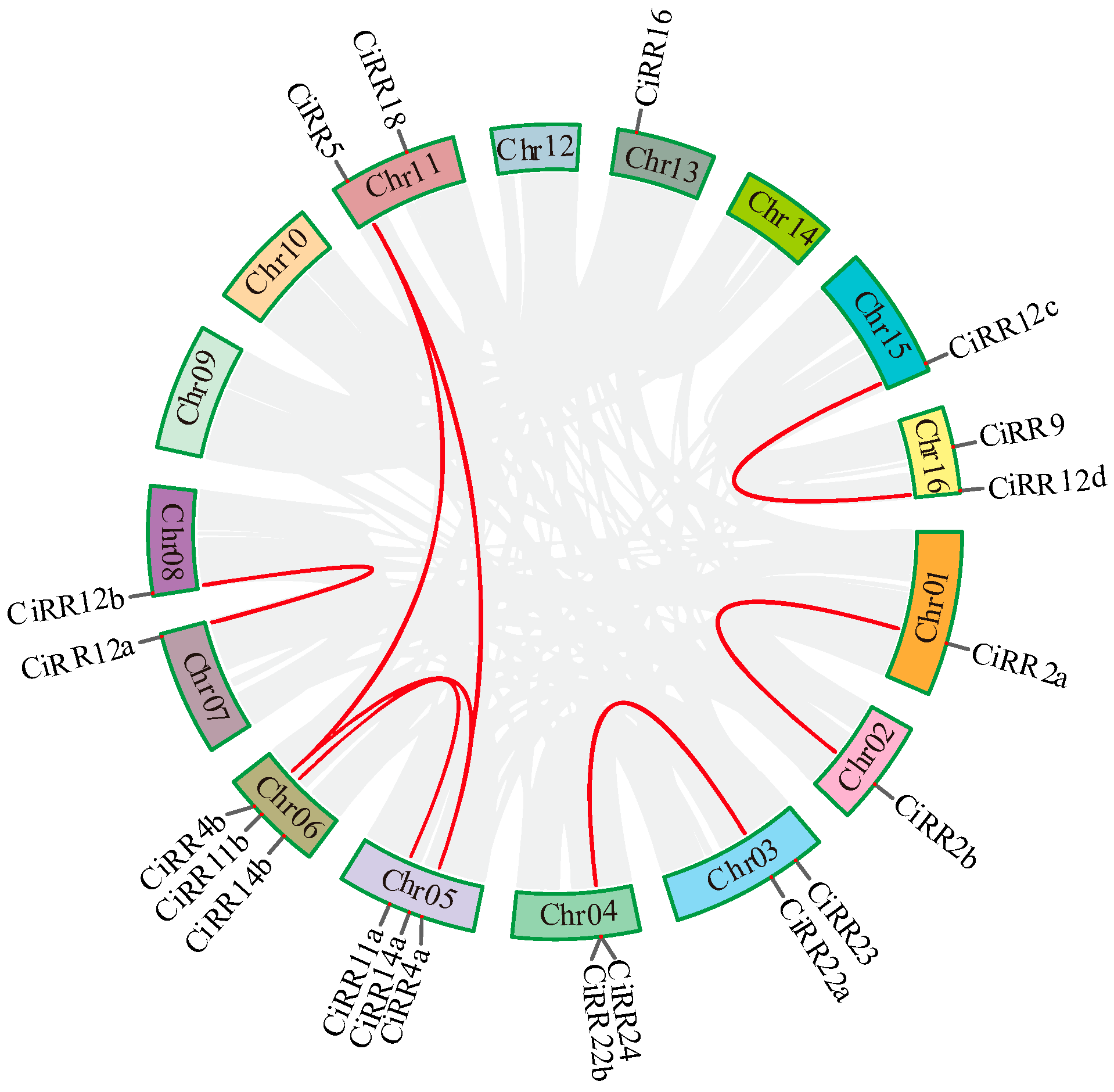
3.3. Phylogenic Relationship and Duplicated Event among CiRRs
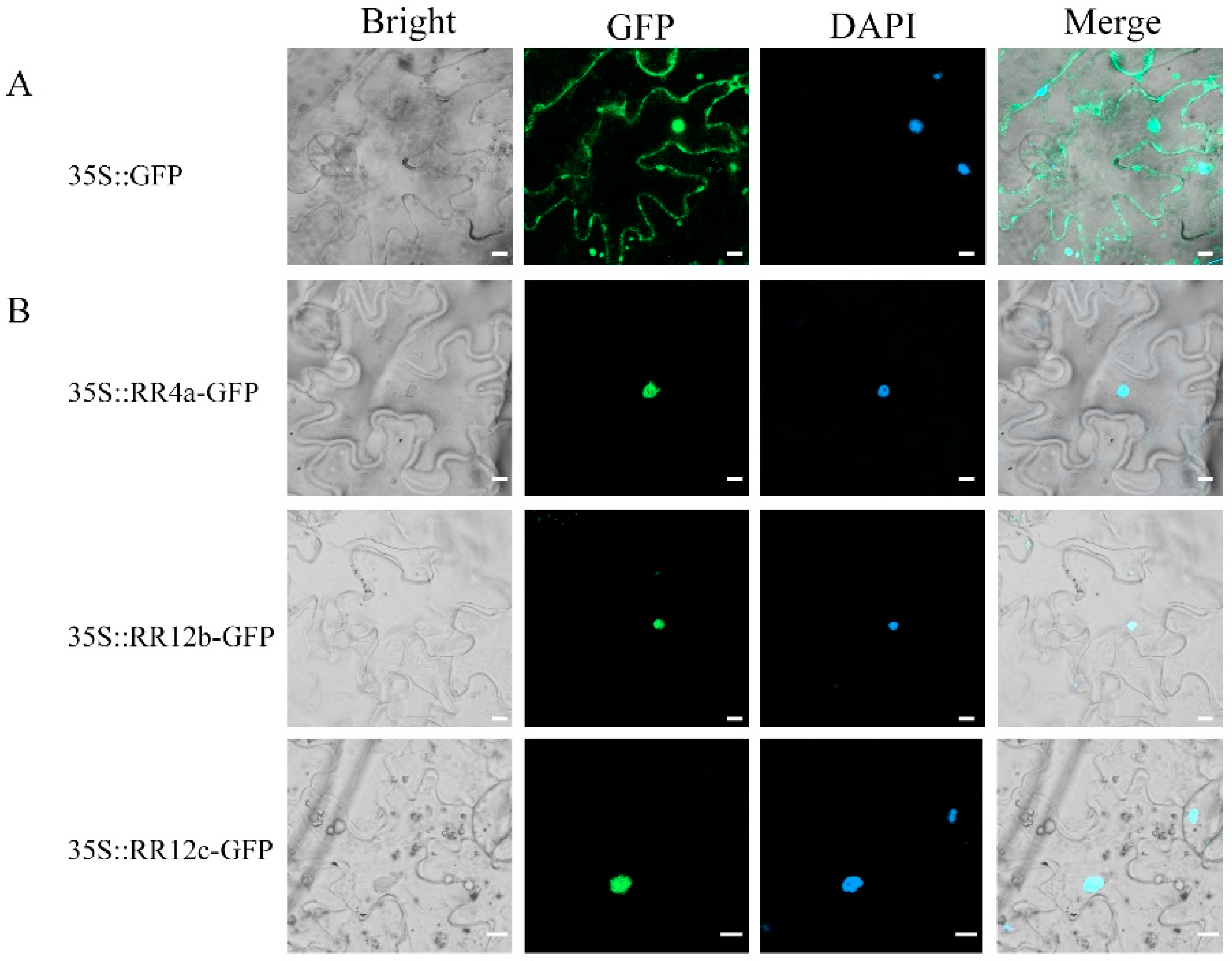
3.4. Structural Characteristics of CiRRs
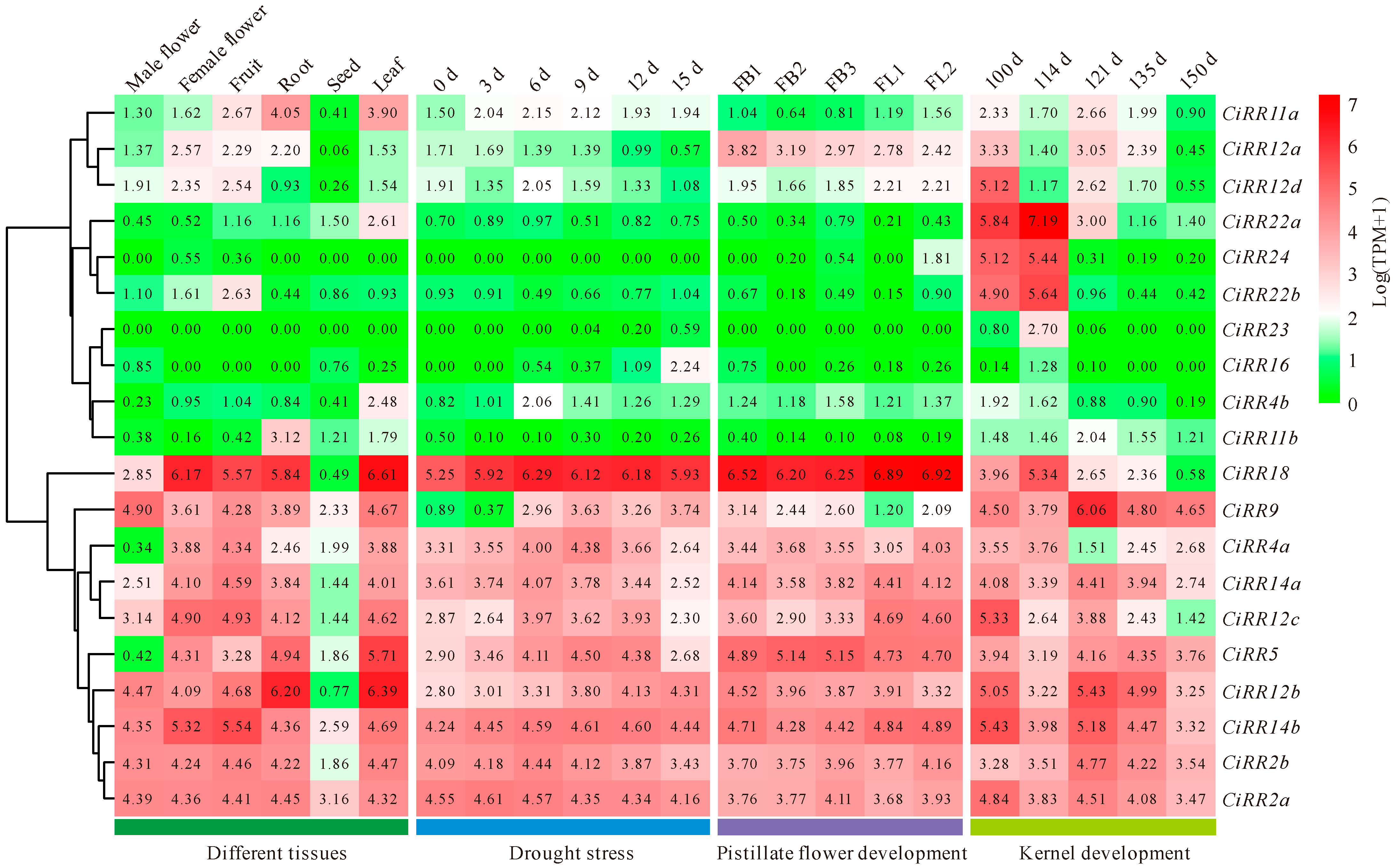
3.5. CiRR Expression Patterns in Various Tissues and during Drought Stress and Kernel Development
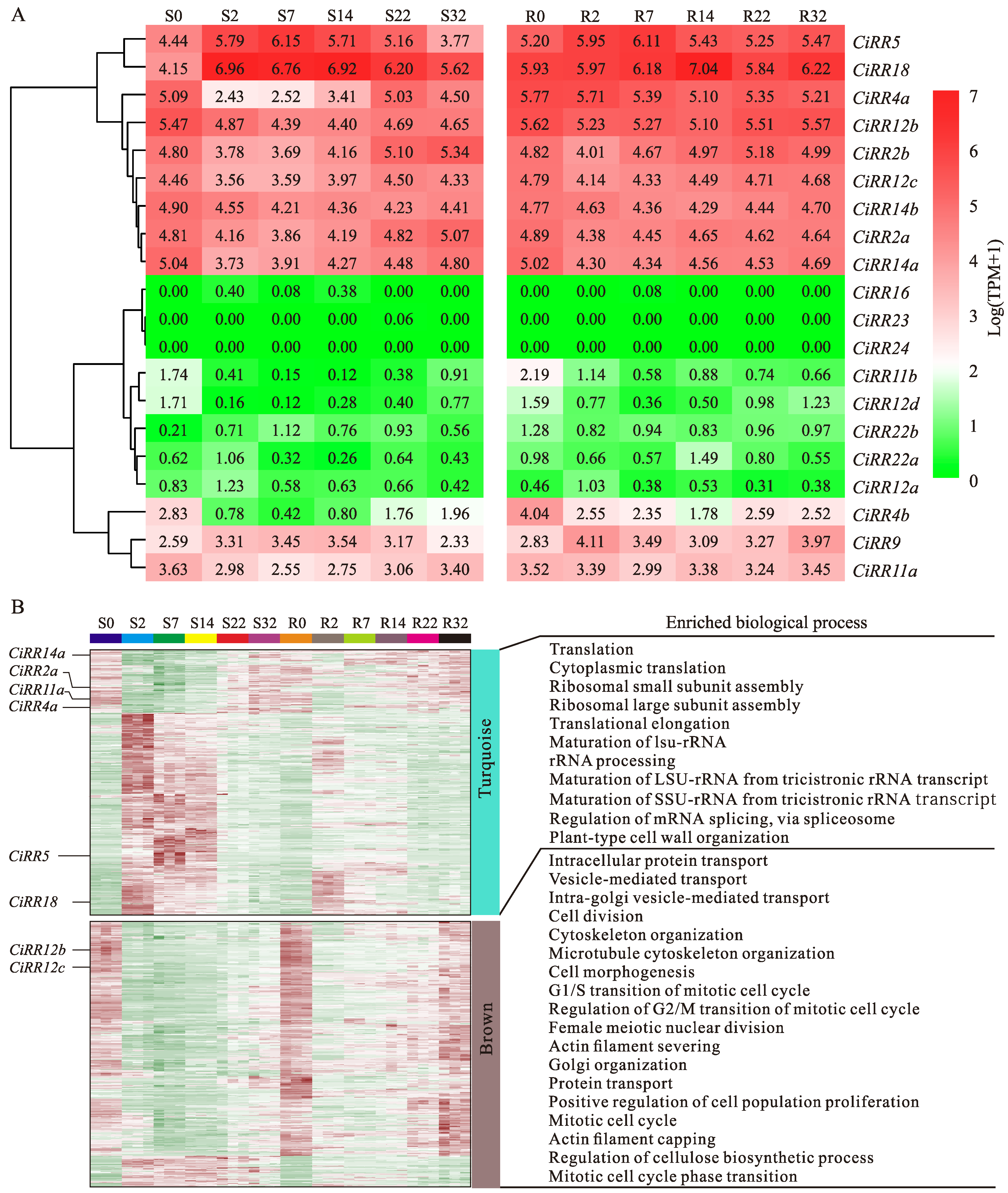
3.6. CiRR Transcriptional Dynamics during Grafting
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cortleven, A.; Leuendorf, J.E.; Frank, M.; Pezzetta, D.; Bolt, S.; Schmülling, T. Cytokinin action in response to abiotic and biotic stresses in plants. Plant Cell Environ. 2019, 42, 998–1018. [Google Scholar] [CrossRef]
- Zwack, P.J.; Rashotte, A.M. Interactions between cytokinin signalling and abiotic stress responses. J. Exp. Bot. 2015, 66, 4863–4871. [Google Scholar] [CrossRef]
- Mo, Z.; Zhang, Y.; Hu, L.; Zhai, M.; Xuan, J. Genome-wide identification and expression analysis of auxin response factor (ARF) gene family in pecan indicates its possible roles during graft union formation. Sci. Hortic. 2023, 322, 112401. [Google Scholar] [CrossRef]
- Mo, Z.; He, H.; Su, W.; Peng, F. Analysis of differentially accumulated proteins associated with graft union formation in pecan (Carya illinoensis). Sci. Hortic. 2017, 224, 126–134. [Google Scholar] [CrossRef]
- Mo, Z.; Jiang, X.; Zhang, Y.; Zhai, M.; Hu, L.; Xuan, J. Weighted gene co-expression network analysis reveals key pathways and hub genes associated with successful grafting in pecan (Carya illinoinensis). Forests 2023, 14, 835. [Google Scholar] [CrossRef]
- Qiu, L.; Jiang, B.; Fang, J.; Shen, Y.; Fang, Z.; Rm, S.K.; Yi, K.; Shen, C.; Yan, D.; Zheng, B. Analysis of transcriptome in hickory (Carya cathayensis), and uncover the dynamics in the hormonal signaling pathway during graft process. BMC Genom. 2016, 17, 935. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Zheng, B. Molecular responses during plant grafting and its regulation by auxins, cytokinins, and gibberellins. Biomolecules 2019, 9, 397. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, Y.; Zhao, M.; Liu, Y.; Xu, X.; Li, T. Transcriptomic analysis of melon/squash graft junction reveals molecular mechanisms potentially underlying the graft union development. PeerJ 2021, 9, e12569. [Google Scholar] [CrossRef] [PubMed]
- Köse, C.; Güleryüz, M. Effects of auxins and cytokinins on graft union of grapevine (Vitis vinifera). N. Z. J. Crop Hort. 2006, 34, 145–150. [Google Scholar] [CrossRef]
- Bidabadi, S.S.; Afazel, M.; Sabbatini, P. Iranian grapevine rootstocks and hormonal effects on graft union, growth and antioxidant responses of Asgari seedless grape. Hortic. Plant J. 2018, 4, 16–23. [Google Scholar] [CrossRef]
- Farsi, M.; Fatahi Moghadam, M.R.; Zamani, Z.; Hassani, D. Effects of scion cultivar, rootstock age and hormonal treatment on minigrafting of Persian walnut. Int. J. Hortic. Sci. Technol. 2018, 5, 185–197. [Google Scholar]
- Cui, Q.; Xie, L.; Dong, C.; Gao, L.; Shang, Q. Stage-specific events in tomato graft formation and the regulatory effects of auxin and cytokinin. Plant Sci. 2021, 304, 110803. [Google Scholar] [CrossRef]
- Ferreira, F.J.; Kieber, J.J. Cytokinin signaling. Curr. Opin. Plant Biol. 2005, 8, 518–525. [Google Scholar] [CrossRef]
- Mira-Rodado, V. New insights into multistep-phosphorelay (MSP)/two-component system (TCS) regulation: Are plants and bacteria that different? Plants 2019, 8, 590. [Google Scholar] [CrossRef]
- Argueso, C.T.; Raines, T.; Kieber, J.J. Cytokinin signaling and transcriptional networks. Curr. Opin. Plant Biol. 2010, 13, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Hou, L.; Meng, J.; You, H.; Li, Z.; Gong, Z.; Yang, S.; Shi, Y. The antagonistic action of abscisic acid and cytokinin signaling mediates drought stress response in Arabidopsis. Mol. Plant 2018, 11, 970–982. [Google Scholar] [CrossRef]
- Ramírez-Carvajal, G.A.; Morse, A.M.; Davis, J.M. Transcript profiles of the cytokinin response regulator gene family in Populus imply diverse roles in plant development. New Phytol. 2008, 177, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Zhao, L.; Mei, J.; Abbas, F.; Xie, X.; Yang, Y.; Huang, Q.; Wang, J.; Yuan, H.; Sharma, A. Genome-wide identification and expression analysis of response regulators family genes in chinese hickory (Carya cathayensis) suggests their potential roles during grafting. J. Plant Growth. Regul. 2023, 42, 5099–5115. [Google Scholar] [CrossRef]
- Hwang, I.; Chen, H.C.; Sheen, J. Two-component signal transduction pathways in Arabidopsis. Plant Physiol. 2002, 129, 500–515. [Google Scholar] [CrossRef]
- Kang, N.Y.; Cho, C.; Kim, J. Inducible expression of Arabidopsis response regulator 22 (ARR22), a type-C ARR, in transgenic Arabidopsis enhances drought and freezing tolerance. PLoS ONE 2013, 8, e79248. [Google Scholar] [CrossRef]
- Ren, B.; Liang, Y.; Deng, Y.; Chen, Q.; Zhang, J.; Yang, X.; Zuo, J. Genome-wide comparative analysis of type-A Arabidopsis response regulator genes by overexpression studies reveals their diverse roles and regulatory mechanisms in cytokinin signaling. Cell Res. 2009, 19, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Zubo, Y.O.; Schaller, G.E. Role of the cytokinin-activated type-B response regulators in hormone crosstalk. Plants 2020, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Ryu, H.; Cho, Y.H.; Scacchi, E.; Sabatini, S.; Hwang, I. Cytokinin-facilitated proteolysis of ARABIDOPSIS RESPONSE REGULATOR 2 attenuates signaling output in two-component circuitry. Plant J. 2012, 69, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Argyros, R.D.; Mathews, D.E.; Chiang, Y.H.; Palmer, C.M.; Thibault, D.M.; Etheridge, N.; Argyros, D.A.; Mason, M.G.; Kieber, J.J.; Schaller, G.E. Type B response regulators of Arabidopsis play key roles in cytokinin signaling and plant development. Plant Cell 2008, 20, 2102–2116. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Yang, X.; Hu, L.; Zhai, M.; Xuan, J. Histological, physio-biochemical, and transcriptomic analyses reveal the potential limiting factors for successful grafting of pecan. J. For. Res. 2024, 35, 35. [Google Scholar] [CrossRef]
- Movahedi, A.; Wei, H.; Pucker, B.; Ghaderi-Zefrehei, M.; Rasouli, F.; Kiani-Pouya, A.; Jiang, T.; Zhuge, Q.; Yang, L.; Zhou, X. Isoprenoid biosynthesis regulation in poplars by methylerythritol phosphate and mevalonic acid pathways. Front. Plant Sci. 2022, 13, 968780. [Google Scholar] [CrossRef]
- Chou, K.C.; Shen, H.B. Plant-mPLoc: A top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE 2010, 5, e11335. [Google Scholar] [CrossRef]
- Zhu, K.; Jin, Q.; Samma, M.K.; Lin, G.; Shen, W. Molecular cloning and characterization of a heme oxygenase1 gene from sunflower and its expression profiles in salinity acclimation. Mol. Biol. Rep. 2014, 41, 4109–4121. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Fan, P.; Liu, H.; Tan, P.; Ma, W.; Mo, Z.; Zhao, J.; Chu, G.; Peng, F. Insight into the CBL and CIPK gene families in pecan (Carya illinoinensis): Identification, evolution and expression patterns in drought response. BMC Plant Biol. 2022, 22, 221. [Google Scholar] [CrossRef]
- Wang, M.; Xi, D.; Chen, Y.; Zhu, C.; Zhao, Y.; Geng, G. Morphological characterization and transcriptome analysis of pistillate flowering in pecan (Carya illinoinensis). Sci. Hortic. 2019, 257, 108674. [Google Scholar] [CrossRef]
- Zhang, C.; Ren, H.; Yao, X.; Wang, K.; Chang, J. Comparative transcriptome analysis reveals differential regulation of flavonoids biosynthesis between kernels of two pecan cultivars. Front. Plant Sci. 2022, 13, 804968. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Shen, L.; Zhao, S.; Li, R.; Song, Y.; Song, S.; Yu, K.; Yang, W.; Li, X.; Sun, J. Genome-wide identification of seed storage protein gene regulators in wheat through coexpression analysis. Plant J. 2021, 108, 1704–1720. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Chen, Y.; Lou, W.; Jia, X.; Zhai, M.; Xuan, J.; Guo, Z.; Li, Y. Identification of suitable reference genes for normalization of real-time quantitative PCR data in pecan (Carya illinoinensis). Trees-Struct. Funct. 2020, 34, 1233–1241. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Iwase, A.; Rymen, B.; Lambolez, A.; Kojima, M.; Takebayashi, Y.; Heyman, J.; Watanabe, S.; Seo, M.; De Veylder, L. Wounding triggers callus formation via dynamic hormonal and transcriptional changes. Plant Physiol. 2017, 175, 1158–1174. [Google Scholar] [CrossRef] [PubMed]
- Nanda, A.K.; Melnyk, C.W. The role of plant hormones during grafting. J. Plant Res. 2018, 131, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, X.; Gao, Y.; Xie, W.; Zhang, L.; Song, J.; Li, S.; Zhao, Z. Genome-wide identification of the RR gene family and its expression analysis in response to TDZ induction in Rhododendron delavayi. Plants 2023, 12, 3250. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, D.; Zhang, L.; Zuo, X.; Fan, S.; Zhang, X.; Shalmani, A.; Han, M. Identification and expression analysis of cytokinin response-regulator genes during floral induction in apple (Malus domestica Borkh). Plant Growth Regul. 2017, 83, 455–464. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, F.; Wang, X.; Feng, J.; Yi, Q.; Zhu, S.; Zhao, X. Mining key genes related to root morphogenesis through genome-wide identification and expression analysis of RR gene family in citrus. Front. Plant Sci. 2022, 13, 1068961. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, Y.; Mi, X.; Gan, L.; Gu, T.; Ding, J.; Li, Y. Identification and expression analysis of cytokinin response regulators in Fragaria vesca. Acta Physiol. Plant. 2016, 38, 198. [Google Scholar] [CrossRef]
- Geng, X.; Zhang, C.; Wei, L.; Lin, K.; Xu, Z.F. Genome-wide identification and expression analysis of cytokinin response regulator (RR) genes in the woody plant Jatropha curcas and functional analysis of JcRR12 in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 11388. [Google Scholar] [CrossRef]
- Qiang, Y.; He, X.; Li, Z.; Li, S.; Zhang, J.; Liu, T.; Tursunniyaz, M.; Wang, X.; Liu, Z.; Fang, L. Genome-wide identification and expression analysis of the response regulator gene family in alfalfa (Medicago sativa L.) reveals their multifarious roles in stress response. Front. Plant Sci. 2023, 14, 1149880. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Li, N.; Zhang, N.; Liu, F.; Wu, J.; Zhao, S.; Shen, J.; Wang, Z.; Peng, L.; Fan, Y. Selective modes affect gene feature and function differentiation of tetraploid Brassica species in their evolution and domestication. Front. Plant Sci. 2023, 14, 1142147. [Google Scholar] [CrossRef] [PubMed]
- Bourret, R.B. Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 2010, 13, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, A.; Paul, L.K.; Sharma, E.; Jha, S.; Jain, M.; Khurana, J.P. OsRR6, a type-A response regulator in rice, mediates cytokinin, light and stress responses when over-expressed in Arabidopsis. Plant Physiol. Bioch. 2021, 161, 98–112. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, F.; Wu, X.; Hu, Y.; Dong, L.; Dewitte, W.; Wen, B. Genome-wide characterization and expression of two-component system genes in cytokinin-regulated gall formation in Zizania latifolia. Plants 2020, 9, 1409. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Shi, B.; Yu, K.; Guo, D.; Ji, X.; Ni, P.; Yang, Y.; Zhang, G.; Yu, Y. Genome-wide characterization of cytokinin response regulator in grape and expression analyses during berry set process. Russ. J. Plant Physl. 2022, 69, 46. [Google Scholar] [CrossRef]
- Zubo, Y.O.; Blakley, I.C.; Yamburenko, M.V.; Worthen, J.M.; Street, I.H.; Franco-Zorrilla, J.M.; Zhang, W.; Hill, K.; Raines, T.; Solano, R. Cytokinin induces genome-wide binding of the type-B response regulator ARR10 to regulate growth and development in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, E5995–E6004. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. 2014, 217, 109–119. [Google Scholar] [CrossRef]
- Kolachevskaya, O.O.; Myakushina, Y.A.; Getman, I.A.; Lomin, S.N.; Deyneko, I.V.; Deigraf, S.V.; Romanov, G.A. Hormonal regulation and crosstalk of auxin/cytokinin signaling pathways in potatoes in vitro and in relation to vegetation or tuberization stages. Int. J. Mol. Sci. 2021, 22, 8207. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Huh, S.U.; Kojima, M.; Sakakibara, H.; Paek, K.H.; Hwang, I. The cytokinin-activated transcription factor ARR2 promotes plant immunity via TGA3/NPR1-dependent salicylic acid signaling in Arabidopsis. Dev. Cell 2010, 19, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Guo, L.; Lu, X.; Malik, W.A.; Zhang, Y.; Wang, J.; Chen, X.; Wang, S.; Wang, J.; Wang, D. Structure and character analysis of cotton response regulator genes family reveals that GhRR7 responses to draught stress. Biol. Res. 2022, 55, 27. [Google Scholar] [CrossRef] [PubMed]
- Zameer, R.; Sadaqat, M.; Fatima, K.; Fiaz, S.; Rasul, S.; Zafar, H.; Qayyum, A.; Nashat, N.; Raza, A.; Shah, A.N. Two-component system genes in Sorghum bicolor: Genome-wide identification and expression profiling in response to environmental stresses. Front. Genet. 2021, 12, 794305. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.A.; Azeem, F.; Afzal, S.; Afzal, N.; Rizwan, M.; Seo, H.; Shah, A.A.; Nawaz, M.A. Comparative omics-based identification and expression analysis of a two-component system in Vigna radiata in drought stress. Agronomy 2023, 13, 989. [Google Scholar] [CrossRef]
- Djeghdir, I.; Chefdor, F.; Bertheau, L.; Koudounas, K.; Carqueijeiro, I.; Cruz, P.L.; Courdavault, V.; Depierreux, C.; Larcher, M.; Lamblin, F. Evaluation of type-B RR dimerization in poplar: A mechanism to preserve signaling specificity? Plant Sci. 2021, 313, 111068. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Shibata, M.; Rymen, B.; Iwase, A.; Bågman, A.M.; Watt, L.; Coleman, D.; Favero, D.S.; Takahashi, T.; Ahnert, S.E. A gene regulatory network for cellular reprogramming in plant regeneration. Plant Cell Physiol. 2018, 59, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Rashotte, A.M.; Carson, S.D.; To, J.P.; Kieber, J.J. Expression profiling of cytokinin action in Arabidopsis. Plant Physiol. 2003, 132, 1998–2011. [Google Scholar] [CrossRef]
- O’Brien, J.A.; Benková, E. Cytokinin cross-talking during biotic and abiotic stress responses. Front. Plant Sci. 2013, 4, 451. [Google Scholar] [CrossRef]
- Hutchison, C.E.; Kieber, J.J. Cytokinin signaling in Arabidopsis. Plant Cell 2002, 14, S47–S59. [Google Scholar] [CrossRef]
- Xie, L.; Dong, C.; Shang, Q. Gene co-expression network analysis reveals pathways associated with graft healing by asymmetric profiling in tomato. BMC Plant Biol. 2019, 19, 373. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, M.; Occhipinti, G.; Principato, G.; Piva, F. Weighted gene co-expression network analysis reveals key genes involved in pancreatic ductal adenocarcinoma development. Cell. Oncol. 2016, 39, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Komaki, S.; Sugimoto, K. Control of the plant cell cycle by developmental and environmental cues. Plant Cell Physiol. 2012, 53, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, H.; Liu, Z.; Li, C.; Wang, R.; Fang, J.; Lu, S.; Guo, J.; Zhu, X.; Wang, X. Integrative analyses of gene expression profile reveal potential crucial roles of mitotic cell cycle and microtubule cytoskeleton in pulmonary artery hypertension. BMC Med. Genom. 2020, 13, 86. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Zhang, T.; Liu, J.; Sun, X.; Sun, X.; Wang, W.; Zheng, C. Interactions between rootstock and scion during grafting and their molecular regulation mechanism. Sci. Hortic. 2023, 308, 111554. [Google Scholar] [CrossRef]
- Iwase, A.; Mitsuda, N.; Koyama, T.; Hiratsu, K.; Kojima, M.; Arai, T.; Inoue, Y.; Seki, M.; Sakakibara, H.; Sugimoto, K. The AP2/ERF transcription factor WIND1 controls cell dedifferentiation in Arabidopsis. Curr. Biol. 2011, 21, 508–514. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Jia, Z.; Wang, G.; Hou, M.; Zhai, M.; Hu, L.; Xuan, J.; Mo, Z. Genome-Wide Identification and Expression Profiling of the Response Regulator (RR) Gene Family in Pecan Reveals Its Possible Association with Callus Formation during Grafting. Forests 2024, 15, 473. https://doi.org/10.3390/f15030473
Zhang Y, Jia Z, Wang G, Hou M, Zhai M, Hu L, Xuan J, Mo Z. Genome-Wide Identification and Expression Profiling of the Response Regulator (RR) Gene Family in Pecan Reveals Its Possible Association with Callus Formation during Grafting. Forests. 2024; 15(3):473. https://doi.org/10.3390/f15030473
Chicago/Turabian StyleZhang, Yan, Zhanhui Jia, Guoming Wang, Mengxin Hou, Min Zhai, Longjiao Hu, Jiping Xuan, and Zhenghai Mo. 2024. "Genome-Wide Identification and Expression Profiling of the Response Regulator (RR) Gene Family in Pecan Reveals Its Possible Association with Callus Formation during Grafting" Forests 15, no. 3: 473. https://doi.org/10.3390/f15030473