
Development of Novel Genomewide Simple Sequence Repeat Markers for Acer truncatum Bunge and Assessment of Their Transferability to Other Closely Related Species


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and DNA Extraction
2.2. SSR Motif Identification
2.3. Unique SSR Screening and Primer Design
2.4. Assessment of SSR Polymorphism
2.5. Cross-Species Transferability
3. Results
3.1. Identification of SSR Loci in Acer Truncatum Genome
3.2. Development of SSR Markers with Specific Physical Positions
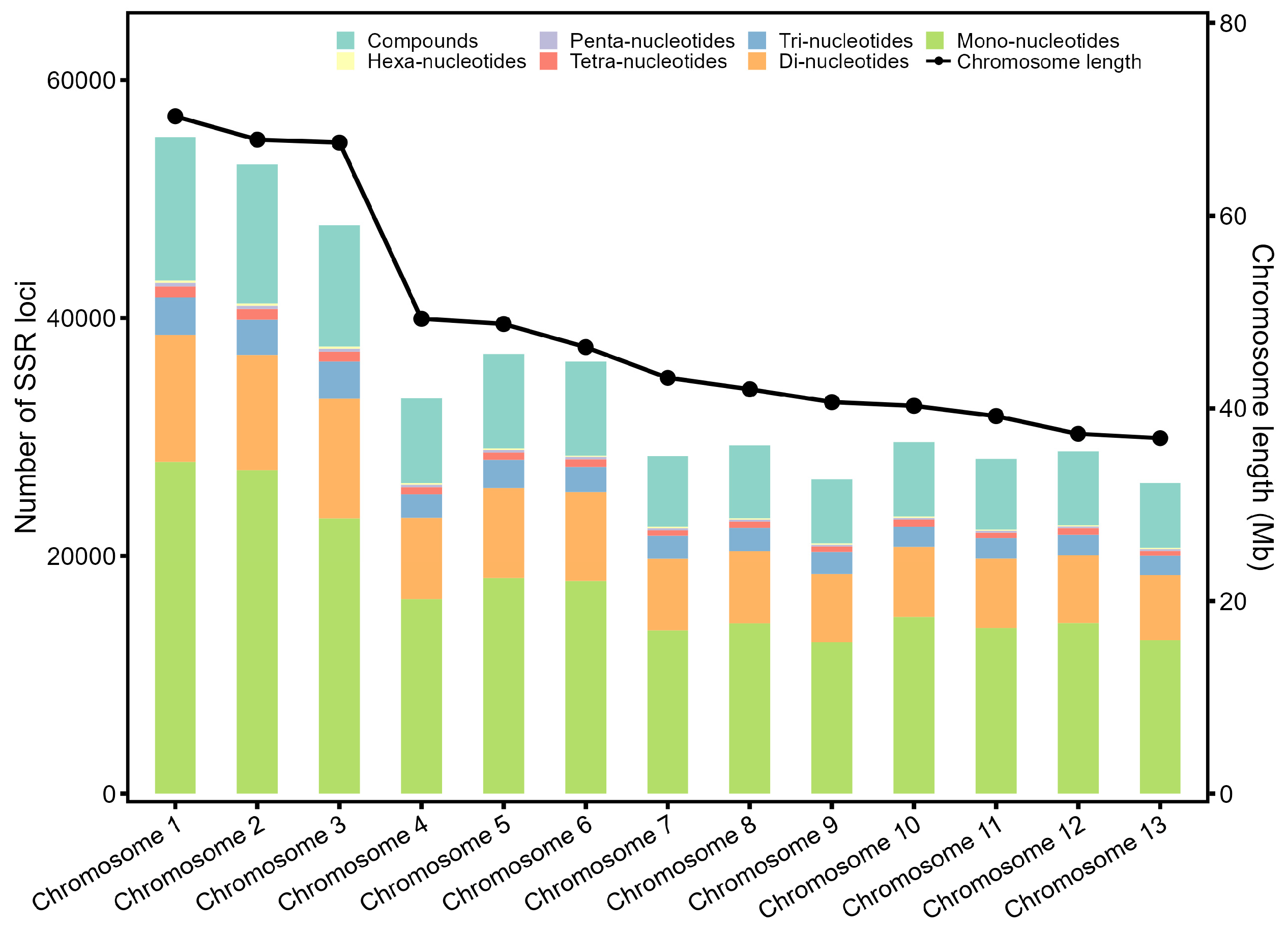
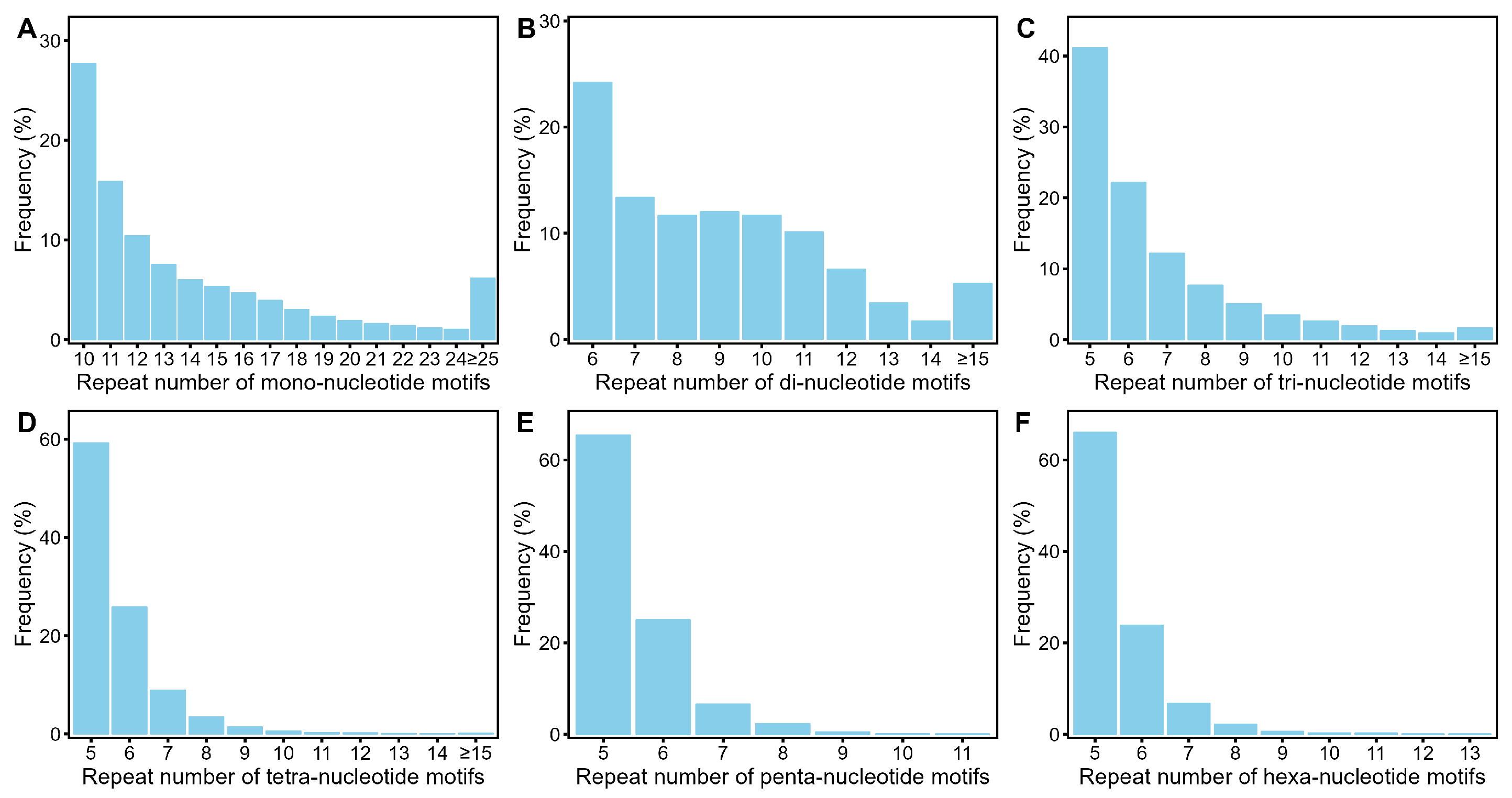
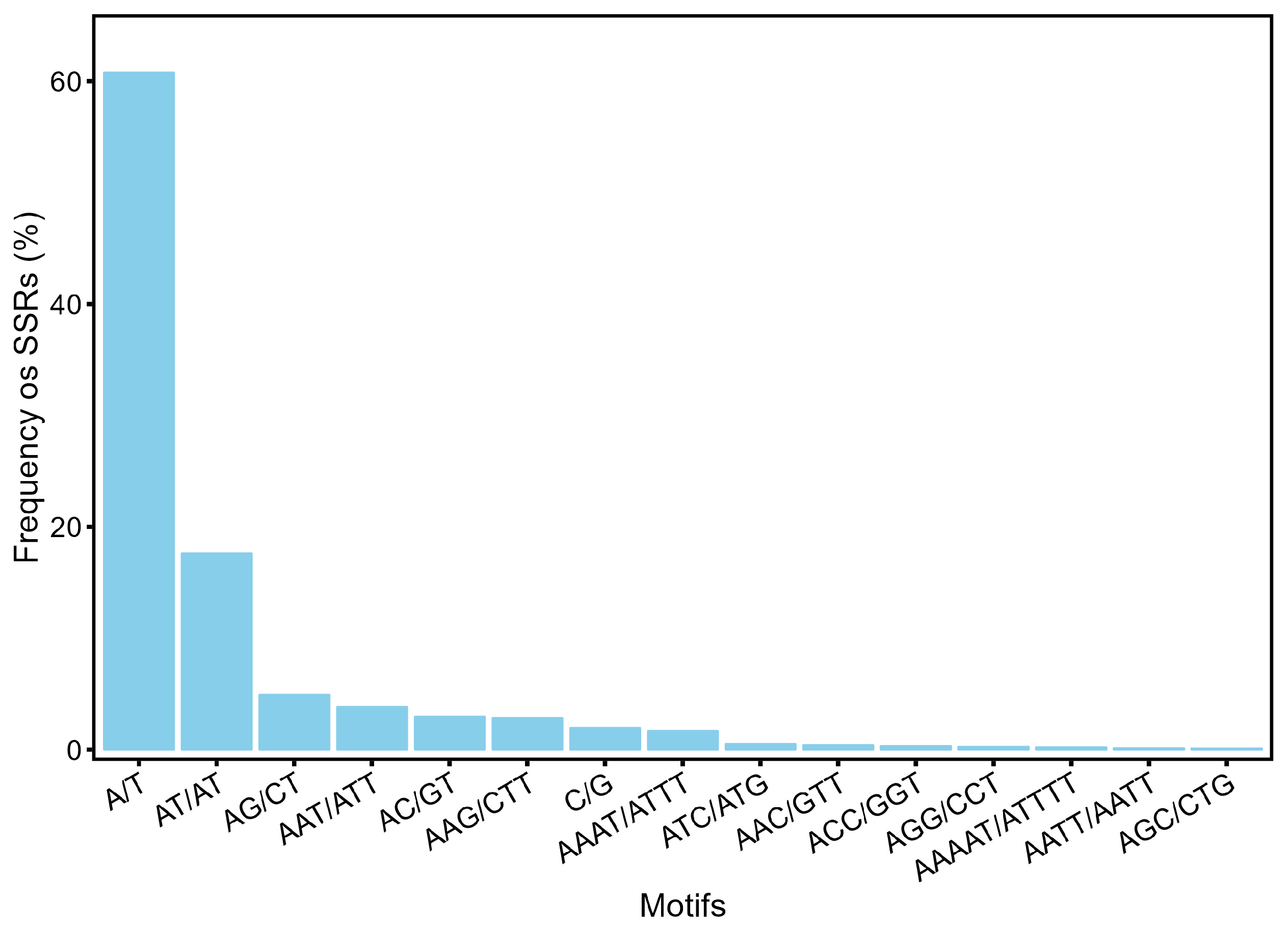
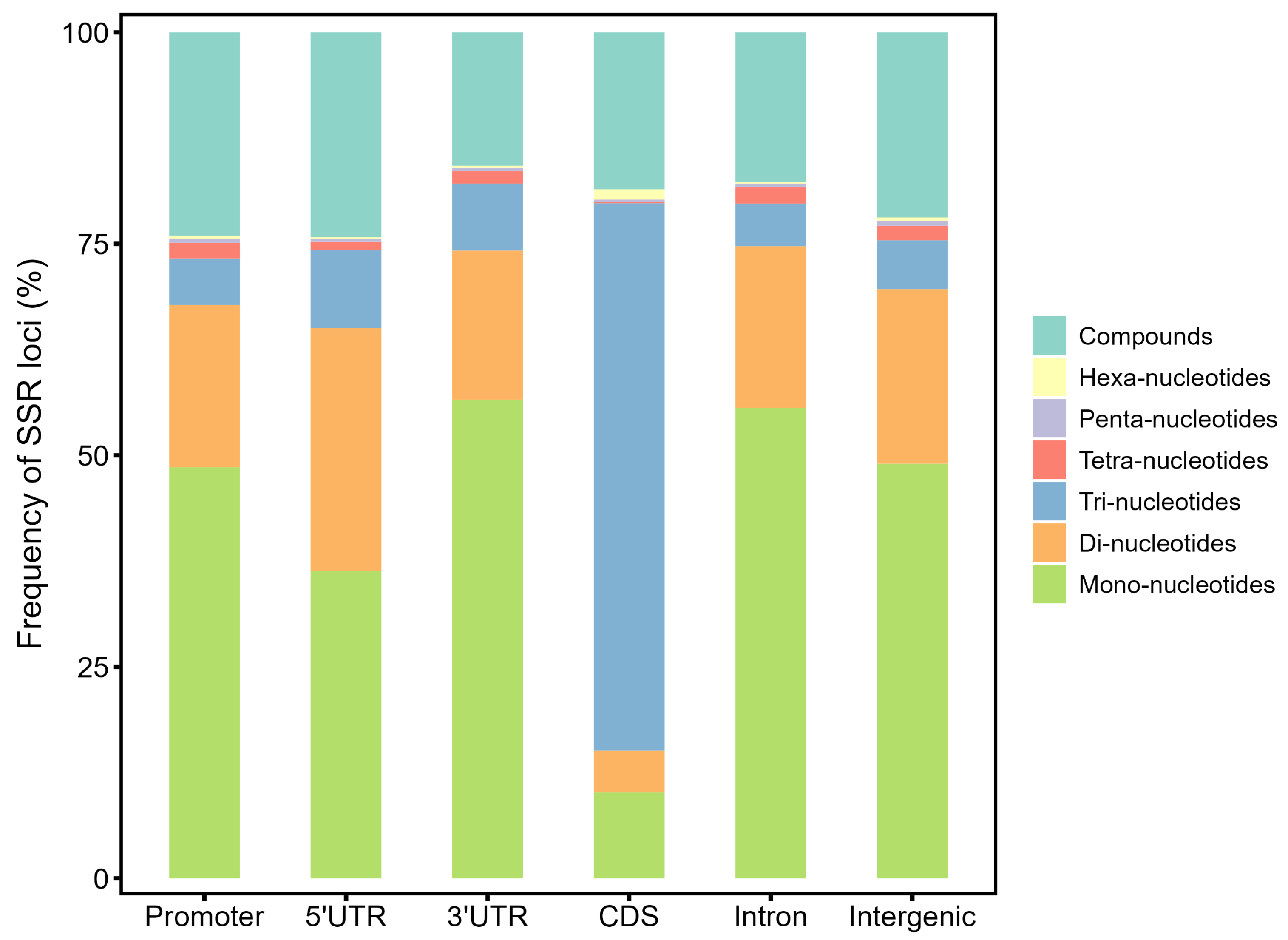
3.3. Genomic Distribution of the SSR Loci
3.4. Validation of SSR Marker Polymorphism
3.5. Cross-Species Transferability Analysis of the SSR Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yi, Y. Application of colored-leaf plants in landscape construction. J. Landsc. Res. 2015, 7, 89–90. [Google Scholar]
- Fan, Y.; Lin, F.; Zhang, R.; Wang, M.; Gu, R.; Long, C. Acer truncatum Bunge: A comprehensive review on ethnobotany, phytochemistry and pharmacology. J. Ethnopharmacol. 2022, 282, 114572. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Wang, W.W.; Yuan, F.L.; Liu, X.; Li, D.L.; Yang, K.Q. Characterization of yuanbaofeng (Acer truncatum Bunge) samaras: Oil, fatty acid, and phytosterol content. Ind. Crop. Prod. 2019, 135, 344–351. [Google Scholar] [CrossRef]
- Tong, Y.; Xv, J.; Chen, F.; Yin, Z.; Liu, N.; Kong, W. Advance in chemical components and products application status of Acer truncatum Bunge. China Oils Fats 2022, 47, 118–123. [Google Scholar] [CrossRef]
- Fan, H.; Sun, L.; Yang, L.; Zhou, J.; Yin, P.; Li, K.; Xue, Q.; Li, X.; Liu, Y. Assessment of the bioactive phenolic composition of Acer truncatum seed coat as a byproduct of seed oil. Ind. Crop. Prod. 2018, 118, 11–19. [Google Scholar] [CrossRef]
- Wang, X.Y.; Fan, J.S.; Wang, S.Y.; Sun, R. A new resource of nervonic acid from purpleblow maple (Acer truncotum) seed oil. For. Prod. J. 2006, 56, 147–150. [Google Scholar]
- Qiao, Q.; Wang, X.; Ren, H.J.; An, K.; Feng, Z.; Cheng, T.T.; Sun, Z.K. Oil content and nervonic acid content of Acer truncatum seeds from 14 regions in China. Hortic. Plant J. 2019, 5, 24–30. [Google Scholar] [CrossRef]
- Ramesh, P.; Mallikarjuna, G.; Sameena, S.; Kumar, A.; Gurulakshmi, K.; Reddy, B.V.; Reddy, P.C.O.; Sekhar, A.C. Advancements in molecular marker technologies and their applications in diversity studies. J. Biosci. 2020, 45, 123. [Google Scholar] [CrossRef]
- Grover, A.; Sharma, P.C. Development and use of molecular markers: Past and present. Crit. Rev. Biotechnol. 2016, 36, 290–302. [Google Scholar] [CrossRef]
- Savadi, S.; Muralidhara, B.M.; Venkataravanappa, V.; Adiga, J.D.; Manjunatha, K.; Patil, B. De novo transcriptome assembly and its utility in development and characterization of the first set of genic SSR markers in cashew. Ind. Crop. Prod. 2022, 189, 115734. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, D.; Bai, L. Development of EST-SSR markers and population genetic structure and genetic diversity of the Malus transitoria (Batalin) C. K. Schneider in Qinghai-Tibetan Plateau. Genet. Resour. Crop. Evol. 2023, 70, 919–933. [Google Scholar] [CrossRef]
- Yan, L.P.; Wang, Y.H.; Wu, D.J.; Ren, F.; Li, Q.H.; Yao, J.X. Optimization and primers screening of SSR-PCR reaction system for Acer truncatum. Mol. Plant Breed. 2021, 19, 2279–2285. [Google Scholar] [CrossRef]
- E, S.T. Taxonomic Study of Acer mono and Acer truncatum in Liaoning Region Based on Phenotype and Molecular Markers; Shenyang Agricultural University: Shenyang, China, 2020. [Google Scholar]
- Wang, R.; Liu, P.; Fan, J.; Li, L. Comparative transcriptome analysis two genotypes of Acer truncatum Bunge seeds reveals candidate genes that influences seed VLCFAs accumulation. Sci. Rep. 2018, 8, 15504. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fan, J.; Chang, P.; Zhu, L.; Zhao, M.; Li, L. Genome survey sequencing of Acer truncatum Bunge to identify genomic information, simple sequence repeat (SSR) markers and complete chloroplast genome. Forests 2019, 10, 87. [Google Scholar] [CrossRef]
- Ma, Q.; Sun, T.; Li, S.; Wen, J.; Zhu, L.; Yin, T.; Yan, K.; Xu, X.; Li, S.; Mao, J.; et al. The Acer truncatum genome provides insights into nervonic acid biosynthesis. Plant J. 2020, 104, 662–678. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, L.; Xu, Y.; Chen, C.; Rong, T.; Ali, F.; Zhou, S.; Wu, F.; Liu, Y.; Wang, J.; et al. Development and characterization of simple sequence repeat markers providing genome-wide coverage and high resolution in maize. DNA Res. 2013, 20, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatic 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin Software. Available online: https://darwin.cirad.fr/ (accessed on 18 January 2024).
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Wang, H.; Gao, S.; Liu, Y.; Wang, P.; Zhang, Z.; Chen, D. A pipeline for effectively developing highly polymorphic simple sequence repeats markers based on multi-sample genomic data. Ecol. Evol. 2022, 12, e8705. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, G.; Shi, A.; Kandel, D.R.; Solís-Gracia, N.; da Silva, J.A.; Avila, C.A. Genome-wide simple sequence repeats (SSR) markers discovered from whole-genome sequence comparisons of multiple spinach accessions. Sci. Rep. 2021, 11, 9999. [Google Scholar] [CrossRef]
- Nonić, M.; Šijačić-Nikolić, M. Genetic diversity: Sources, threats, and conservation. In Life on Land; Leal Filho, W., Azul, A.M., Brandli, L., Lange Salvia, A., Wall, T., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 421–435. [Google Scholar]
- Soltis, P.S.; Soltis, D.E. Plant genomes: Markers of evolutionary history and drivers of evolutionary change. Plants People Planet 2021, 3, 74–82. [Google Scholar] [CrossRef]
- Carvalho, Y.G.S.; Vitorino, L.C.; Souza, U.J.B.d.; Bessa, L.A. Recent trends in research on the genetic diversity of plants: Implications for conservation. Diversity 2019, 11, 62. [Google Scholar] [CrossRef]
- Ma, Y.; Devi, M.J.; Song, L.; Gao, H.; Cao, B. Genome survey sequencing of Ailanthus altissima and identification of simple sequence repeat (SSR) markers. Silvae Genet. 2022, 71, 47–53. [Google Scholar] [CrossRef]
- Bansal, S.; Kumar, A.; Lone, A.A.; Khan, M.H.; Malhotra, E.V.; Singh, R. Development of novel genome-wide simple sequence repeats (SSR) markers in Bunium persicum. Ind. Crop. Prod. 2022, 178, 114625. [Google Scholar] [CrossRef]
- Hong, W.; Hao, Z.; Liu, K.; Luo, H.; Bi, R.; Yuan, Z.; Zong, S.; Wang, J. Development and identification of SSR molecular markers based on whole genomic sequences of Punica granatum. J. Beijing For. Univ. 2019, 41, 38–47. [Google Scholar] [CrossRef]
- Liu, C.; Li, J.; Qin, G. Genome-wide distribution of simple sequence repeats in pomegranate and their application to the analysis of genetic diversity. Tree Genet. 2020, 16, 36. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, G.; Wu, W.R. Characterization and comparison of microsatellites in Gramineae. Genom. Appl. Biol. 2011, 30, 513–520. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Gunter, L.E.; Yang, Z.-K.; Yin, T.; Sewell, M.M.; DiFazio, S.P. Characterization of microsatellites revealed by genomic sequencing of Populus trichocarpa. Can. J. For. Res. 2004, 34, 85–93. [Google Scholar] [CrossRef]
- Gur-Arie, R.; Cohen, C.J.; Eitan, Y.; Shelef, L.; Hallerman, E.M.; Kashi, Y. Simple sequence repeats in Escherichia coli: Abundance, distribution, composition, and polymorphism. Genome Res. 2000, 10, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.J.; Zhang, L. Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol. 2006, 7, R14. [Google Scholar] [CrossRef] [PubMed]
- La Rota, M.; Kantety, R.V.; Yu, J.K.; Sorrells, M.E. Nonrandom distribution and frequencies of genomic and EST-derived microsatellite markers in rice, wheat, and barley. BMC Genom. 2005, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.L.; Santini, L.; Diniz, A.L.; Munhoz Cde, F. Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biol. 2016, 39, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Ren, X.; Mason, A.S.; Li, J.; Wang, W.; Xiao, M.; Fu, D. Revisiting an important component of plant genomes: Microsatellites. Funct. Plant Biol. 2013, 40, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Deng, Y.; Tan, J.; Hu, S.; Yu, J.; Xue, Q. A genome-wide microsatellite polymorphism database for the indica and japonica rice. DNA Res. 2007, 14, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Du, F.K.; Xu, F.; Qu, H.; Feng, S.; Tang, J.; Wu, R. Exploiting the transcriptome of Euphrates Poplar, Populus euphratica (Salicaceae) to develop and characterize new EST-SSR markers and construct an EST-SSR database. PLoS ONE 2013, 8, e61337. [Google Scholar] [CrossRef] [PubMed]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Singh, A.; Choudhary, S.; Bhardwaj, P. Transcriptome characterization and development of functional polymorphic SSR marker resource for Himalayan endangered species, Taxus contorta (Griff). Ind. Crop. Prod. 2019, 140, 111600. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Tian, X.; Strassmann, J.E.; Queller, D.C. Genome nucleotide composition shapes variation in simple sequence repeats. Mol. Biol. Evol. 2010, 28, 899–909. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Repeat Units | All SSR Loci | SSR Loci with Unique Flanking Sequences | e-PCR-Screened Specific SSR Loci | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number | Length (bp) | Rate (%) | Number | Length (bp) | Rate (%) | Number | Length (bp) | Rate (%) | ||
| Perfect SSRs | MNRs | 229,106 | 14.54 | 49.55 | 141,466 | 14.89 | 49.40 | 103,001 | 14.90 | 51.50 |
| DNRs | 93,700 | 18.41 | 20.27 | 56,354 | 19.29 | 19.68 | 42,226 | 19.21 | 21.11 | |
| TNRs | 28,355 | 19.67 | 6.13 | 15,601 | 20.32 | 5.45 | 12,230 | 20.04 | 6.12 | |
| TTRs | 8049 | 22.62 | 1.74 | 5250 | 22.73 | 1.83 | 3873 | 22.71 | 1.94 | |
| PNRs | 2530 | 27.38 | 0.55 | 1618 | 27.37 | 0.57 | 1147 | 27.17 | 0.57 | |
| HNRs | 1742 | 32.83 | 0.38 | 1088 | 32.85 | 0.38 | 696 | 32.64 | 0.35 | |
| Total | 363,482 | 16.29 | 78.62 | 221,377 | 16.76 | 77.31 | 163,173 | 16.75 | 81.59 | |
| Compound SSRs | - | 98,849 | 93.70 | 21.38 | 64,967 | 93.91 | 22.69 | 36,817 | 77.33 | 18.41 |
| Total | - | 462,331 | 32.85 | 100.00 | 286,344 | 34.26 | 100.00 | 199,990 | 27.90 | 100.00 |
| Genomic Regions | All SSR Loci | SSR Loci with Unique Flanking Sequences | e-PCR-Screened Specific SSR Loci | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | Density (SSRs/Mbp) | Length (bp) | Rate (%) | Number | Density (SSRs/Mbp) | Length (bp) | Rate (%) | Number | Density (SSRs/Mbp) | Length (bp) | Rate (%) | |
| Promotor | 61,431 | 1247.6 | 35.26 | 13.16 | 45,802 | 930.2 | 35.34 | 15.80 | 32,848 | 667.1 | 29.36 | 16.19 |
| 5′UTR | 4334 | 1555.8 | 35.92 | 0.93 | 3661 | 1314.2 | 36.41 | 1.26 | 3477 | 1248.2 | 32.47 | 1.71 |
| 3′UTR | 3706 | 733.8 | 26.33 | 0.79 | 3103 | 614.4 | 26.26 | 1.07 | 2837 | 561.7 | 24.74 | 1.40 |
| CDS | 3525 | 111.5 | 34.90 | 0.76 | 2708 | 85.7 | 35.41 | 0.93 | 2534 | 80.2 | 31.06 | 1.25 |
| Intron | 55,307 | 793.5 | 28.67 | 11.85 | 38,419 | 551.2 | 28.87 | 13.25 | 31,541 | 452.5 | 24.56 | 15.55 |
| Intergenic | 338,497 | 706.6 | 33.53 | 72.51 | 196,229 | 409.6 | 35.68 | 67.68 | 129,658 | 270.7 | 28.73 | 63.90 |
| Total */average | 466,800 | 732.3 | 33.16 | 100.00 | 289,922 | 454.8 | 34.63 | 100.00 | 202,895 | 318.3 | 28.22 | 100.00 |
| SSR Type | Number | Monomorphic in Acer truncatum Plus Trees | Polymorphic in Acer truncatum Plus Trees | Transferability | Polymorphic in All Species a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Acer henryi | Acer tataricum subsp. ginnala | Acer negundo | Acer palmatum | Acer grosseri | Acer platanoides | Acer rubrum | |||||
| MNR | 15 | 2 (13.3 b) | 10 (66.7) | 9 (60.0) | 8 (53.3) | 9 (60.0) | 9 (60.0) | 9 (60.0) | 10 (66.7) | 10 (66.7) | 7 (46.7) |
| DNR | 15 | 2 (13.3) | 9 (60.0) | 6 (40.0) | 6 (40.0) | 5 (33.3) | 7 (46.7) | 4 (26.7) | 7 (46.7) | 5 (33.3) | 3 (20.0) |
| TNR | 15 | 2 (13.3) | 12 (80.0) | 10 (66.7) | 10 (66.7) | 8 (53.3) | 10 (66.7) | 8 (53.3) | 12 (80.0) | 10 (66.7) | 7 (46.7) |
| TTR | 15 | 2 (13.3) | 13 (86.7) | 10 (66.7) | 10 (66.7) | 10 (66.7) | 10 (66.7) | 10 (66.7) | 11 (73.3) | 10 (66.7) | 6 (40.0) |
| PNR | 15 | 0 | 12 (80.0) | 5 (33.3) | 6 (40.0) | 6 (40.0) | 6 (40.0) | 6 (40.0) | 7 (46.7) | 6 (40.0) | 4 (26.7) |
| HNR | 15 | 1 (6.7) | 11 (73.3) | 3 (20.0) | 2 (13.3) | 3 (20.0) | 5 (33.3) | 2 (13.3) | 3 (20.0) | 5 (33.3) | 1 (6.7) |
| Compound | 15 | 0 | 15 (100.0) | 13 (86.7) | 11 (73.3) | 11 (73.3) | 12 (80.0) | 12 (80.0) | 12 (80.0) | 13 (86.7) | 8 (53.3) |
| Total | 105 | 9 (8.6) | 82 (78.1) | 56 (53.3) | 53 (50.5) | 52 (49.5) | 59 (56.2) | 51 (48.6) | 62 (59.0) | 59 (56.2) | 36 (34.3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, Y.; Bao, H.; Jin, Q.; Wu, Z.; Han, M.; Zhang, C.; Wang, J. Development of Novel Genomewide Simple Sequence Repeat Markers for Acer truncatum Bunge and Assessment of Their Transferability to Other Closely Related Species. Forests 2024, 15, 635. https://doi.org/10.3390/f15040635
Long Y, Bao H, Jin Q, Wu Z, Han M, Zhang C, Wang J. Development of Novel Genomewide Simple Sequence Repeat Markers for Acer truncatum Bunge and Assessment of Their Transferability to Other Closely Related Species. Forests. 2024; 15(4):635. https://doi.org/10.3390/f15040635
Chicago/Turabian StyleLong, Yixin, Hasengaowa Bao, Qingyu Jin, Zhiping Wu, Minghai Han, Chi Zhang, and Jun Wang. 2024. "Development of Novel Genomewide Simple Sequence Repeat Markers for Acer truncatum Bunge and Assessment of Their Transferability to Other Closely Related Species" Forests 15, no. 4: 635. https://doi.org/10.3390/f15040635
APA StyleLong, Y., Bao, H., Jin, Q., Wu, Z., Han, M., Zhang, C., & Wang, J. (2024). Development of Novel Genomewide Simple Sequence Repeat Markers for Acer truncatum Bunge and Assessment of Their Transferability to Other Closely Related Species. Forests, 15(4), 635. https://doi.org/10.3390/f15040635