

Genetic Divergence and Evolutionary Adaption of Four Wild Almond Species (Prunus spp. L.)



Abstract
:1. Introduction
2. Materials and Methods
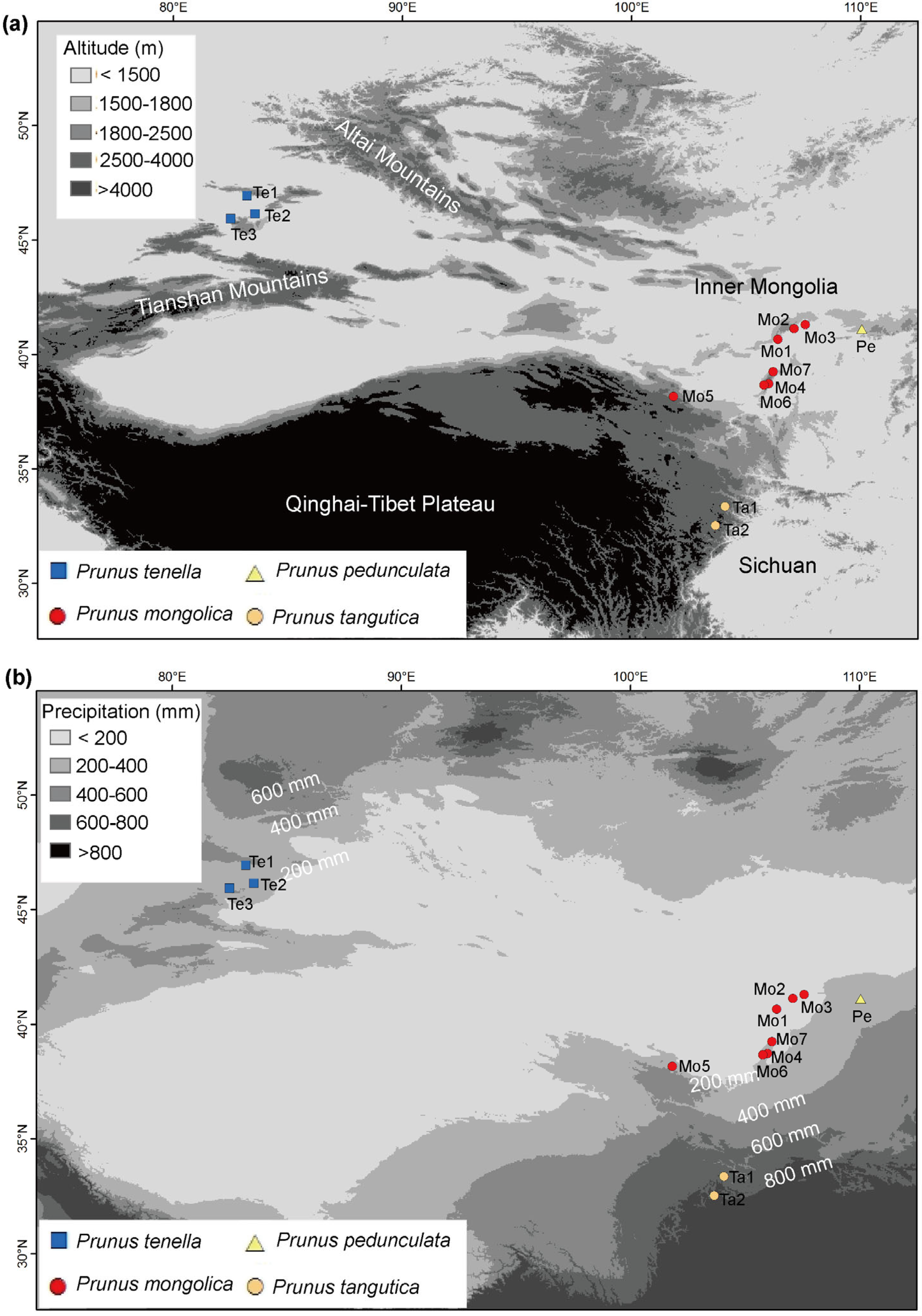
2.1. Plant Sampling and DNA Extraction
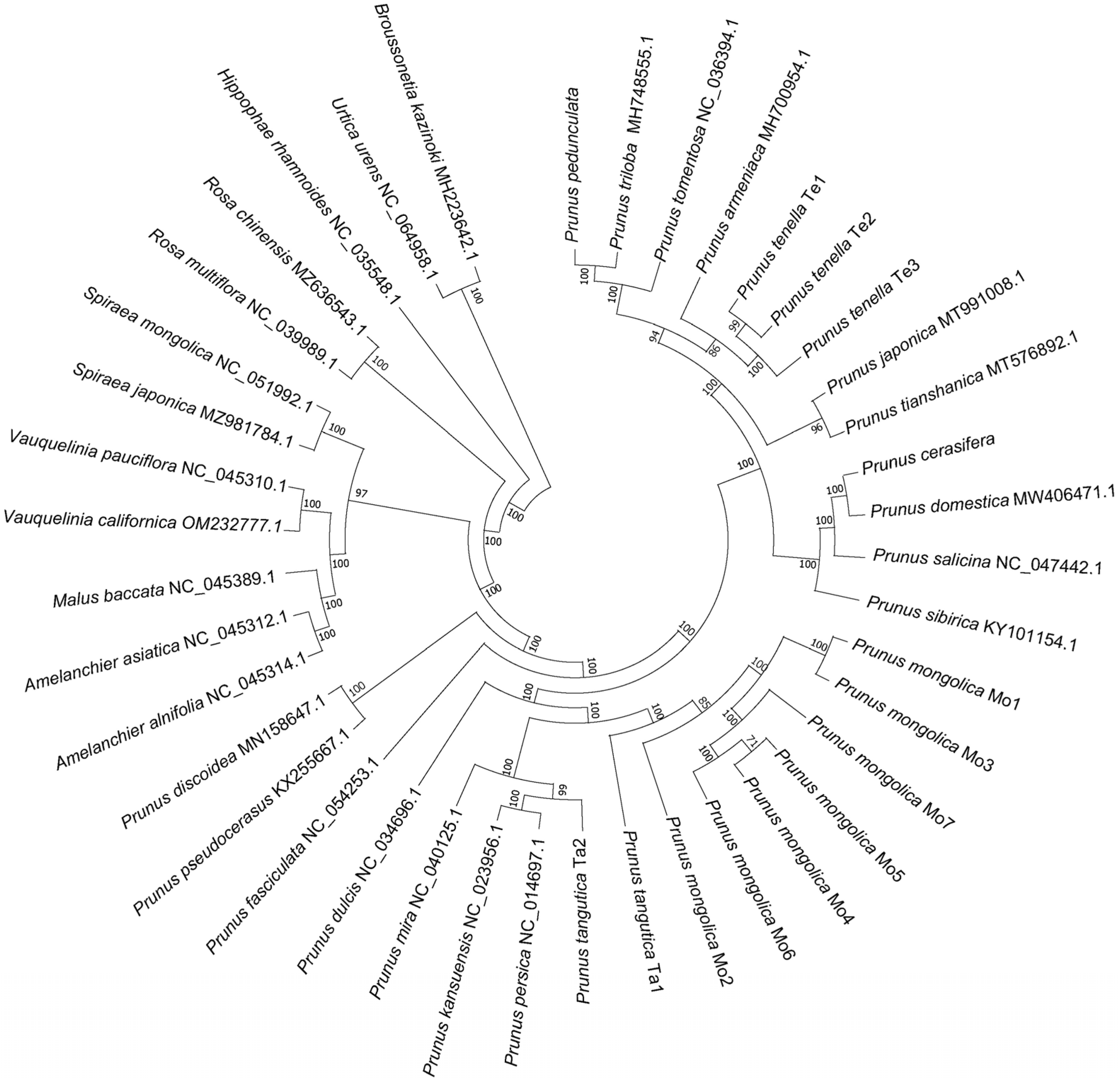
2.2. Sequencing, Chloroplast Genome Assembly and Phylogenetic Construction
2.3. RAD Sequencing and SNP Calling
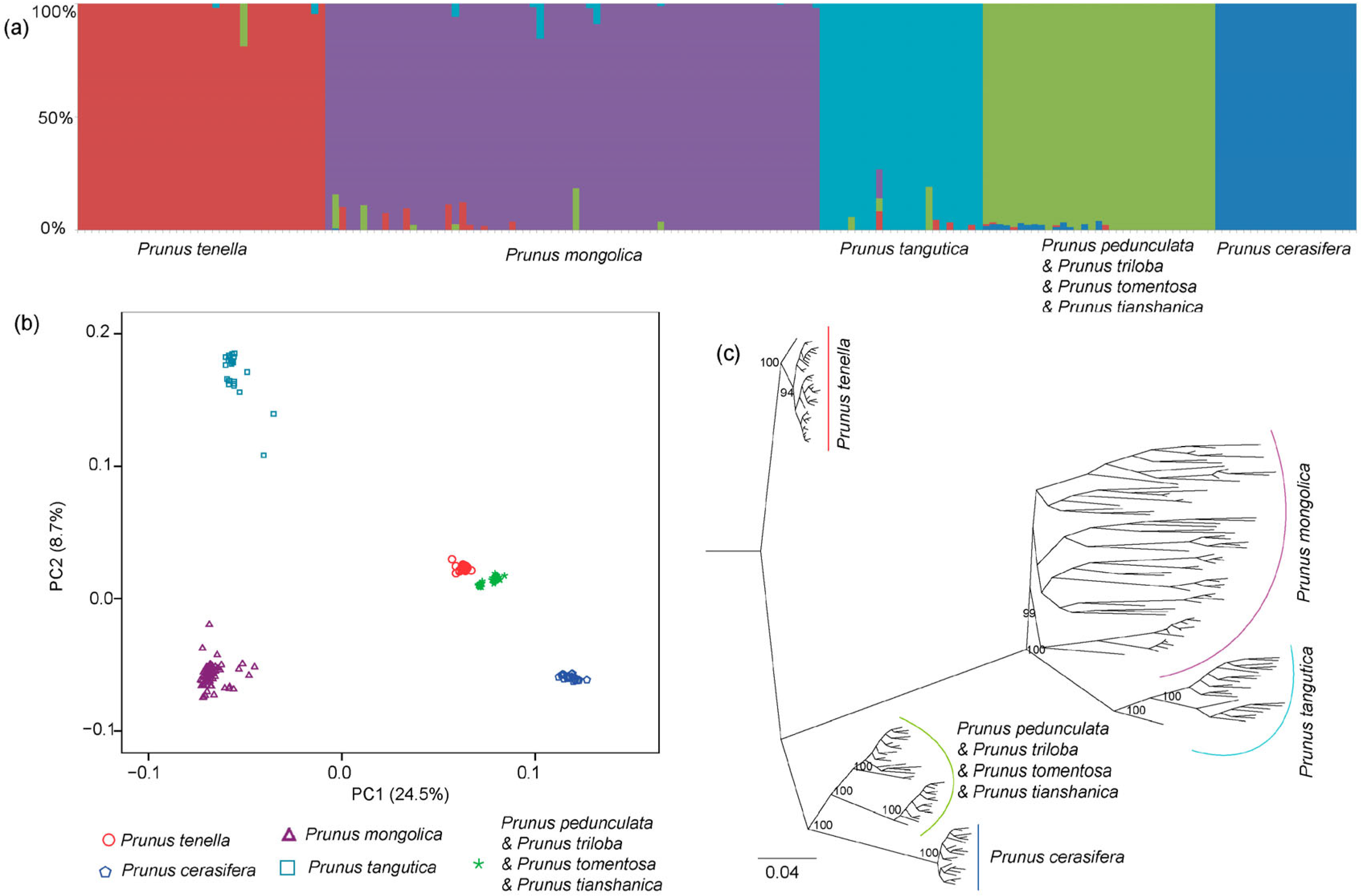
2.4. Genetic Structure Analyses
2.5. Demographical History Analysis
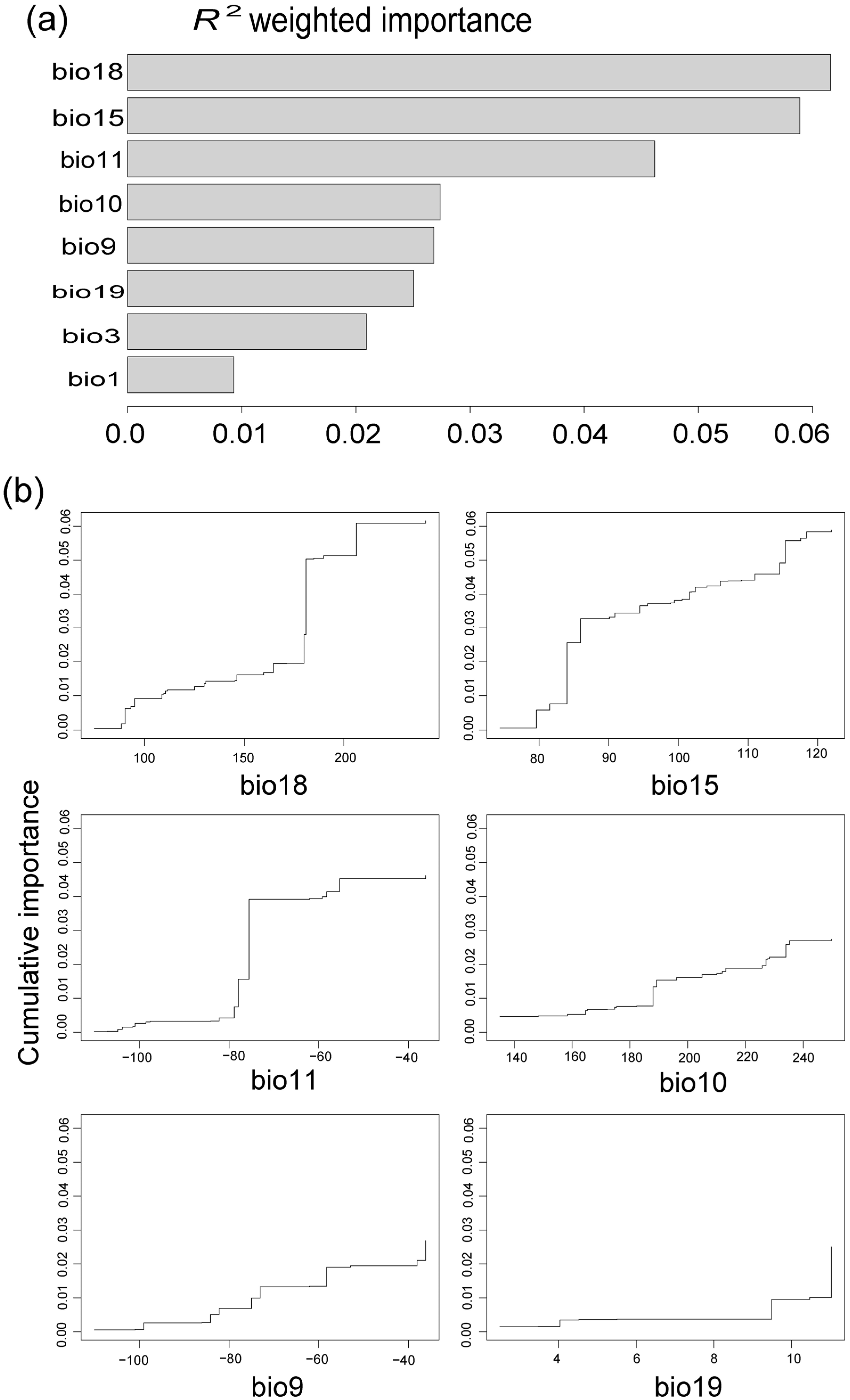
2.6. Genetic Environment Association Analysis
2.7. Potential Loci Related to Local Adaptation
3. Results
3.1. Chloroplast Phylogeny
3.2. Genetic Structure and Demographical History
3.3. Genetic Environment Association
4. Discussion
4.1. Origin of Polyploid P. pedunculata and P. triloba
4.2. Evolution of P. tenella
4.3. Local Adaptation of P. mongolica to Arid Environment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Stern, D.L. The genetic causes of convergent evolution. Nat. Rev. Genet. 2013, 14, 751–764. [Google Scholar] [CrossRef]
- Elmer, K.R.; Meyer, A. Adaptation in the age of ecological genomics: Insights from parallelism and convergence. Trends Ecol. Evol. 2011, 26, 298–306. [Google Scholar] [CrossRef]
- Oke, K.B.; Rolshausen, G.; LeBlond, C.; Hendry, A.P. How parallel is parallel evolution? A comparative analysis in fishes. Am. Nat. 2017, 190, 1–16. [Google Scholar] [CrossRef]
- Roda, F.; Ambrose, L.; Walter, G.M.; Liu, H.L.; Schaul, A.; Lowe, A.; Pelser, P.B.; Prentis, P.; Rieseberg, L.H.; Ortiz-Barrientos, D. Genomic evidence for the parallel evolution of coastal forms in the Senecio lautus complex. Mol. Ecol. 2013, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Duran-Castillo, M.; Hudson, A.; Wilson, Y.; Field, D.L.; Twyford, A.D. A phylogeny of Antirrhinum reveals parallel evolution of alpine morphology. New Phytol. 2022, 233, 1426–1439. [Google Scholar] [CrossRef]
- Fan, L.-L.; Tang, L.-S.; Wu, L.-F.; Ma, J.; Li, Y. The limited role of snow water in the growth and development of ephemeral plants in a cold desert. J. Veg. Sci. 2014, 25, 681–690. [Google Scholar] [CrossRef]
- Zhou, H.; Zhao, W.; Zhang, G. Varying water utilization of Haloxylon ammodendron plantations in a desert-oasis ecotone. Hydrol. Process. 2017, 31, 825–835. [Google Scholar] [CrossRef]
- Song, C.; Hu, S.; Han, W.; Zhang, T.; Fang, X.; Gao, J.; Wu, F. Middle Miocene to earliest Pliocene sedimentological and geochemical records of climate change in the western Qaidam Basin on the NE Tibetan Plateau. Palaeogeogr. Palaeoclim. Palaeoecol. 2014, 395, 67–76. [Google Scholar] [CrossRef]
- Tang, Z.-H.; Ding, Z.-L. A palynological insight into the Miocene aridification in the Eurasian interior. Palaeoworld 2013, 22, 77–85. [Google Scholar] [CrossRef]
- Yang, X.; Scuderi, L.; Paillou, P.; Liu, Z.; Li, H.; Ren, X. Quaternary environmental changes in the drylands of China—A critical review. Quat. Sci. Rev. 2011, 30, 3219–3233. [Google Scholar] [CrossRef]
- Guan, Q.; Pan, B.; Li, N.; Zhang, J.; Xue, L. Timing and significance of the initiation of present day deserts in the northeastern Hexi Corridor, China. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2011, 306, 70–74. [Google Scholar]
- Zhang, M.-L.; Fritsch, P.W. Evolutionary response of Caragana (Fabaceae) to Qinghai-Tibetan Plateau uplift and Asian interior aridification. Plant Syst. Evol. 2010, 288, 191–199. [Google Scholar] [CrossRef]
- Yao, G.-Q.; Nie, Z.-F.; Turner, N.C.; Li, F.-M.; Gao, T.-P.; Fang, X.-W.; Scoffoni, C. Combined high leaf hydraulic safety and efficiency provides drought tolerance in Caragana species adapted to low mean annual precipitation. New Phytol. 2021, 229, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Ladizinsky, G. On the origin of almond. Genet. Resour. Crop Evol. 1999, 46, 143–147. [Google Scholar] [CrossRef]
- Yazbek, M.M.; Al-Zein, M.S. Wild almonds gone wild: Revisiting Darwin’s statement on the origin of peaches. Genet. Resour. Crop Evol. 2014, 61, 1319–1328. [Google Scholar] [CrossRef]
- Vander Wall, S.B. The evolutionary ecology of nut dispersal. Bot. Rev. 2001, 67, 74–117. [Google Scholar] [CrossRef]
- Wang, W.; Yang, T.; Wang, H.-L.; Li, Z.-J.; Ni, J.-W.; Su, S.; Xu, X.-Q. Comparative and phylogenetic analyses of the complete chloroplast genomes of six almond species (Prunus spp. L.). Sci. Rep. 2020, 10, 10137. [Google Scholar] [CrossRef]
- Chin, S.-W.; Shaw, J.; Haberle, R.; Wen, J.; Potter, D. Diversification of almonds, peaches, plums and cherries—Molecular systematics and biogeographic history of Prunus (Rosaceae). Mol. Phylogenet. Evol. 2014, 76, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Yazbek, M.; Oh, S.H. Peaches and almonds: Phylogeny of Prunus subg. Amygdalus (Rosaceae) based on DNA sequences and morphology. Plant Syst. Evol. 2013, 299, 1403–1418. [Google Scholar] [CrossRef]
- Fernandez-Mazuecos, M.; Mellers, G.; Vigalondo, B.; Saez, L.; Vargas, P.; Glover, B.J. Resolving recent plant radiations: Power and robustness of Genotyping-by-Sequencing. Syst. Biol. 2018, 67, 250–268. [Google Scholar] [CrossRef]
- Twyford, A.D.; Ennos, R.A. Next-generation hybridization and introgression. Heredity 2012, 108, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Srivastav, M.; Clement, W.L.L.; Landrein, S.; Zhang, J.; Howarth, D.G.G.; Donoghue, M.J.J. A phylogenomic analysis of Lonicera and its bearing on the evolution of organ fusion. Am. J. Bot. 2023, 110, e16143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-X.; Wang, Q.; Wen, Z.-B. Spatial genetic structure of Prunus mongolica in arid Northwestern China based on RAD sequencing data. Diversity 2021, 13, 397. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Collin, F.-D.; Durif, G.; Raynal, L.; Lombaert, E.; Gautier, M.; Vitalis, R.; Marin, J.-M.; Estoup, A. Extending approximate Bayesian computation with supervised machine learning to infer demographic history from genetic polymorphisms using DIYABC Random Forest. Mol. Ecol. Resour. 2021, 21, 2598–2613. [Google Scholar] [CrossRef]
- Yu, Y.; Fu, J.; Xu, Y.; Zhang, J.; Ren, F.; Zhao, H.; Tian, S.; Guo, W.; Tu, X.; Zhao, J.; et al. Genome re-sequencing reveals the evolutionary history of peach fruit edibility. Nat. Commun. 2018, 9, 5404. [Google Scholar] [CrossRef]
- Gugger, P.F.; Liang, C.T.; Sork, V.L.; Hodgskiss, P.; Wright, J.W. Applying landscape genomic tools to forest management and restoration of Hawaiian koa (Acacia koa) in a changing environment. Evol. Appl. 2018, 11, 231–242. [Google Scholar] [CrossRef]
- Jiang, X.-L.; Gardner, E.M.; Meng, H.-H.; Deng, M.; Xu, G.-B. Land bridges in the Pleistocene contributed to flora assembly on the continental islands of South China: Insights from the evolutionary history of Quercus championii. Mol. Phylogenet. Evol. 2019, 132, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Hill, D.J.; Dolan, A.M.; Carnaval, A.C.; Haywood, A.M. PaleoClim, high spatial resolution paleoclimate surfaces for global land areas. Sci. Data 2018, 5, 180254. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan v2.6: Community Ecology Package. Available online: https://CRAN.R-project.org/package=vegan (accessed on 5 March 2024).
- Frichot, E.; Francois, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genome wide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Capblancq, T.; Luu, K.; Blum, M.G.B.; Bazin, E. Evaluation of redundancy analysis to identify signatures of local adaptation. Mol. Ecol. Resour. 2018, 18, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Su, G. Chromosome numbers of six species in the genus Amygdalus from China. J. Wuhan Bot. Res. 1985, 3, 363–366. [Google Scholar]
- Wan, T.; Qiao, B.-X.; Zhou, J.; Shao, K.-S.; Pan, L.-Y.; An, F.; He, X.-S.; Liu, T.; Li, P.-K.; Cai, Y.-L. Evolutionary and phylogenetic analyses of 11 Cerasus species based on the complete chloroplast genome. Front. Plant Sci. 2023, 14, 1070600. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Shi, G.; Zhen, L.; Wang, X. Karyotype parameters analysis and genetic relationship discussion of Cerasus (Rosaceae). J. Nanjing For. Univ. Nat. Sci. Ed. 2014, 38, 25–29. [Google Scholar]
- Hodel, R.G.J.; Zimmer, E.; Wen, J. A phylogenomic approach resolves the backbone of Prunus (Rosaceae) and identifies signals of hybridization and allopolyploidy. Mol. Phylogenet. Evol. 2021, 160, 107118. [Google Scholar] [CrossRef] [PubMed]
- Torfstein, A.; Steinberg, J. The Oligo-Miocene closure of the Tethys Ocean and evolution of the proto-Mediterranean Sea. Sci. Rep. 2020, 10, 13817. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-P.; Li, H.-Q.; Zhao, L.; Man, L.; Liu, Q. Mapping the ecological dimensions and potential distributions of endangered relic shrubs in western Ordos biodiversity center. Sci. Rep. 2016, 6, 26268. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, R.; Bai, S.; Gao, X.; Liu, M.; Yan, W. Mongolian Almond (Prunus mongolica Maxim): The morpho-physiological, biochemical and transcriptomic response to drought stress. PLoS ONE 2015, 10, e0124442. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, E.; Andreu, P.; Fillat, M.F.; Luisa Peleato, M.; Marin, J.A.; Arbeloa, A. Identification of early salt-stress-responsive proteins in In Vitro Prunus cultured excised roots. Plants 2022, 16, 2101. [Google Scholar] [CrossRef]
- Li, S.; Zheng, G.; Wang, F.; Yu, H.; Wang, S.; Guan, H.; Lv, F.; Xia, Y. Expression and Functional Analysis of the PaPIP1-2 Gene during Dormancy and Germination Periods of Kernel-Using Apricot (Prunus armeniaca L.). Forests 2023, 14, 2306. [Google Scholar] [CrossRef]
- Jimenez, S.; Dridi, J.; Gutierrez, D.; Moret, D.; Irigoyen, J.J.; Moreno, M.A.; Gogorcena, Y. Physiological, biochemical and molecular responses in four Prunus rootstocks submitted to drought stress. Tree Physiol. 2013, 33, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Yu, W.; Yang, X.; Liang, J.; Sun, X.; Sun, M.; Xiao, Y.; Peng, F. Silicon enhances the drought resistance of peach seedlings by regulating hormone, amino acid, and sugar metabolism. BMC Plant Biol. 2022, 22, 422. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Luo, Q.; Tian, Y.; Meng, F. Physiological and proteomic analyses of the drought stress response in Amygdalus Mira (Koehne) Yu et Lu roots. BMC Plant Biol. 2017, 17, 53. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Population Code | Location | Voucher Specimen | Nind |
|---|---|---|---|---|
| Prunus tenella | ||||
| Te1 | Tacheng, Xinjiang, China | AN-TC-01 | 11 | |
| Te2 | Tuoli, Xinjiang, China | AN-TL-01 | 12 | |
| Te3 | Yumin, Xinjiang, China | AN-YM-01 | 12 | |
| Prunus mongolica | ||||
| Mo1 | Dengkou, Inner Mongolia, China | AM-DK-001 | 14 | |
| Mo2 | Wulatehou Banner, Inner Mongolia, China | AM-WHQ-001 | 12 | |
| Mo3 | Wulate Middle Banner, Inner Mongolia, China | AM-WZQ-001 | 1 | |
| Mo4 | Yinchuan, Ningxia, China | AM-YC-001 | 12 | |
| Mo5 | Yongchang, Gansu, China | AM-YCX-001 | 8 | |
| Mo6 | Alxa Left Banner, Inner Mongolia, China | AM-ZQG-001 | 13 | |
| Mo7 | Alxa Left Banner, Inner Mongolia, China | AM-ZQZ-001 | 10 | |
| Prunus tangutica | ||||
| Ta1 | Jiuzhaigou, Sichuan, China | AX-JZG-01 | 15 | |
| Ta2 | Songpan, Sichuan, China | AX-SP-01 | 8 | |
| Prunus pedunculata | Pe | Guyang, Inner Mongolia, China | AC-BT-01 | 12 |
| Prunus triloba | Botanical Garden in Urumqi, Xinjiang, China | — | 4 | |
| Prunus tomentosa | Helan Mountain, Ningxia, China | MYT-YC-01 | 2 | |
| Prunus tianshanica | Tekes, Xinjiang, China | CT-TKS-01 | 15 | |
| Prunus cerasifera | Huocheng, Xinjiang, China | PC-HC-01 | 20 |
| Parameter | Expectation | 95% CI |
|---|---|---|
| N1 | 3.3 × 105 | 2.9 × 103–1.1 × 106 |
| N2 | 8.5 × 106 | 1.2 × 105–2.6 × 107 |
| N3 | 4.9 × 105 | 7.8 × 102–2.3 × 106 |
| N4 | 7.0 × 106 | 3.5 × 104–9.3 × 106 |
| N5 | 4.1 × 106 | 1.2 × 102–2.2 × 107 |
| t1 | 2.93 Ma | 0.03–8.95 Ma |
| t2 | 2.36 Ma | 0.01–8.58 Ma |
| t3 | 7.81 Ma | 0.05–27.97 Ma |
| t4 | 17.77 Ma | 0.20–47.29 Ma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.-X.; Zhang, X.-F.; Zhang, J. Genetic Divergence and Evolutionary Adaption of Four Wild Almond Species (Prunus spp. L.). Forests 2024, 15, 834. https://doi.org/10.3390/f15050834
Zhang H-X, Zhang X-F, Zhang J. Genetic Divergence and Evolutionary Adaption of Four Wild Almond Species (Prunus spp. L.). Forests. 2024; 15(5):834. https://doi.org/10.3390/f15050834
Chicago/Turabian StyleZhang, Hong-Xiang, Xiao-Fang Zhang, and Jian Zhang. 2024. "Genetic Divergence and Evolutionary Adaption of Four Wild Almond Species (Prunus spp. L.)" Forests 15, no. 5: 834. https://doi.org/10.3390/f15050834
APA StyleZhang, H.-X., Zhang, X.-F., & Zhang, J. (2024). Genetic Divergence and Evolutionary Adaption of Four Wild Almond Species (Prunus spp. L.). Forests, 15(5), 834. https://doi.org/10.3390/f15050834