

Alien Species Introduction and Demographic Changes Contributed to the Population Genetic Structure of the Nut-Yielding Conifer Torreya grandis (Taxaceae)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. Locus Amplification and Sequencing
2.3. Analyses of Genetic Diversity and Population Structure
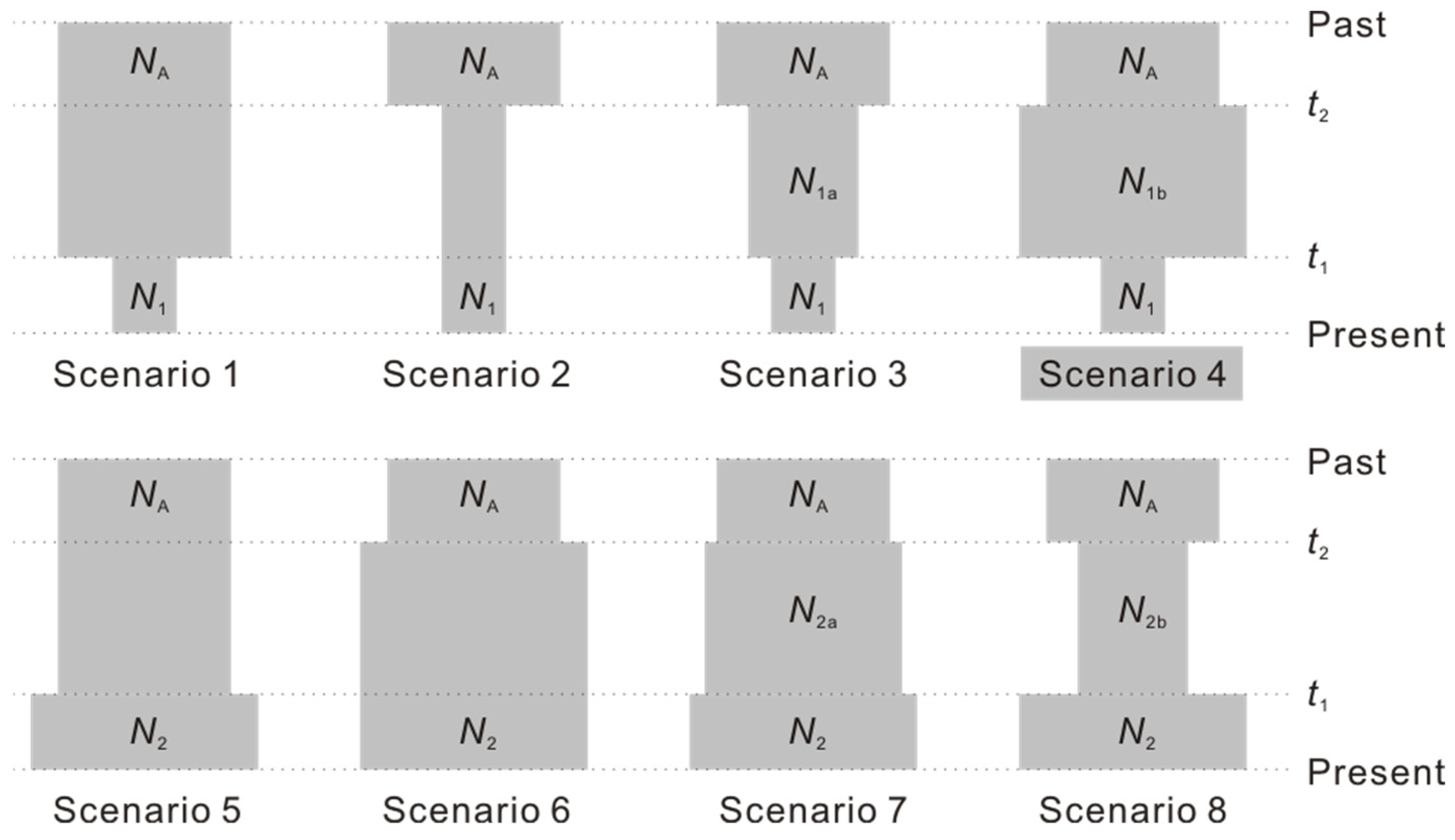
2.4. Inference of Demographic History
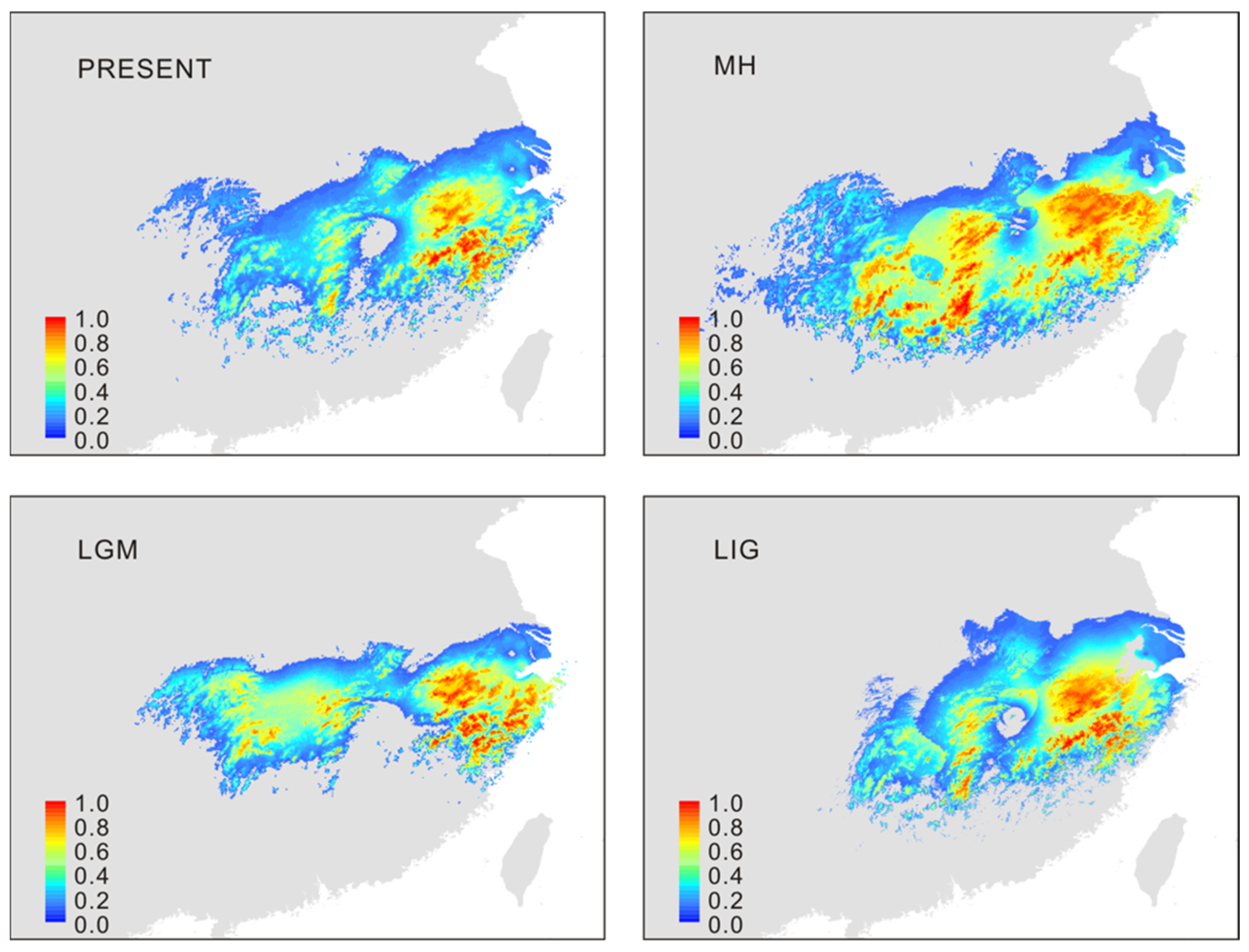
2.5. Ecological Niche Modeling
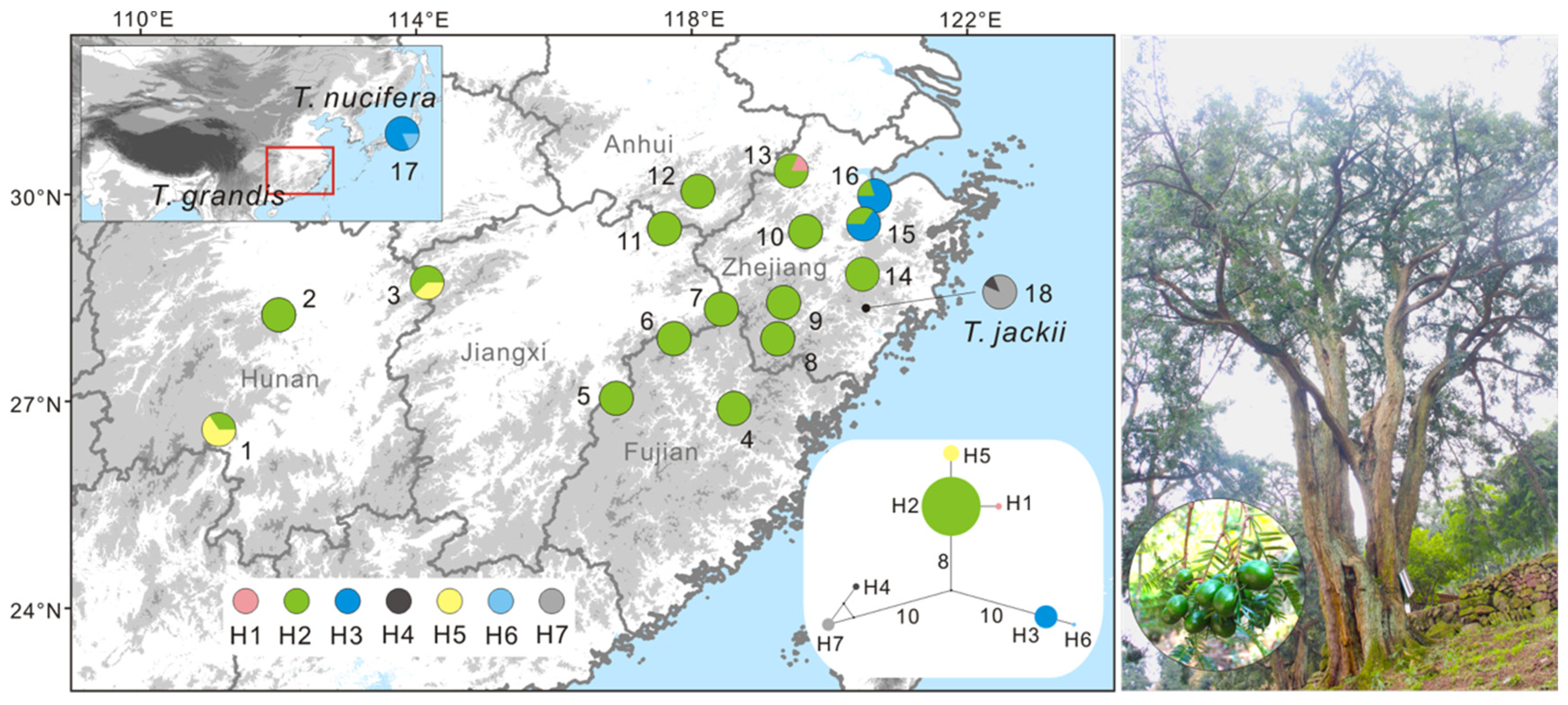
3. Results
3.1. Genetic Diversity
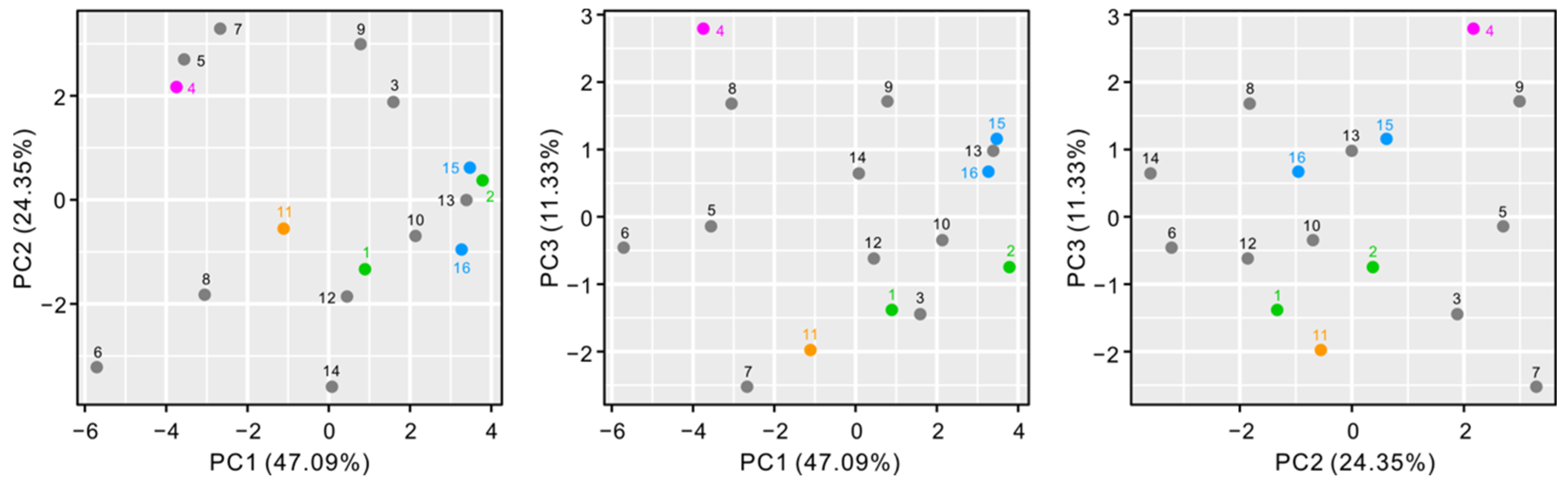
3.2. Population Genetic Structure
3.3. Demographic History
3.4. Projected Distribution with Niche Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Foll, M.; Gaggiotti, O. Identifying the environmental factors that determine the genetic structure of populations. Genetics 2006, 174, 875–891. [Google Scholar] [CrossRef] [PubMed]
- Ogwu, M.C.; Osawaru, M.E.; Ahana, C.M. Challenges in conserving and utilizing plant genetic resources (PGR). Int. J. Genet. Mol. Biol. 2014, 6, 16–23. [Google Scholar] [CrossRef]
- Govindaraj, M.; Vetriventhan, M.; Srinivasan, M. Importance of genetic diversity assessment in crop plants and its recent advances: An overview of its analytical perspectives. Genet. Res. Int. 2015, 2015, 431487. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. B 2004, 359, 183–195. [Google Scholar] [CrossRef]
- Hamrick, J.L. Response of forest trees to global environmental changes. For. Ecol. Manag. 2004, 197, 323–335. [Google Scholar] [CrossRef]
- Jump, A.S.; Penuelas, J. Running to stand still: Adaptation and the response of plants to rapid climate change. Ecol. Lett. 2005, 8, 1010–1020. [Google Scholar] [CrossRef]
- Willi, Y.; Van Buskirk, J.; Schmid, B.; Fischer, M. Genetic isolation of fragmented populations is exacerbated by drift and selection. J. Evol. Biol. 2007, 20, 534–542. [Google Scholar] [CrossRef]
- Lee, C.R.; Mitchell-Olds, T. Quantifying effects of environmental and geographical factors on patterns of genetic differentiation. Mol. Ecol. 2011, 20, 4631–4642. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef] [PubMed]
- Bradburd, G.S.; Ralph, P.L.; Coop, G.M. Disentangling the effects of geographic and ecological isolation on genetic differentiation. Evolution 2013, 67, 3258–3273. [Google Scholar] [CrossRef]
- Fu, Y.B. Understanding crop genetic diversity under modern plant breeding. Theor. Appl. Genet. 2015, 128, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Van Nocker, S.; Gardiner, S.E. Breeding better cultivars, faster: Applications of new technologies for the rapid deployment of superior horticultural tree crops. Hortic. Res. 2014, 1, 14022. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, Y. Natural hybridization, introgression breeding, and cultivar improvement in the genus Actinidia. Tree Genet. Genomes 2014, 10, 1113–1122. [Google Scholar] [CrossRef]
- Jia, Y.; Milne, R.I.; Zhu, J.; Gao, L.M.; Zhu, G.F.; Zhao, G.F.; Liu, J.; Li, Z.H. Evolutionary legacy of a forest plantation tree species (Pinus armandii): Implications for widespread afforestation. Evol. Appl. 2020, 13, 2646–2662. [Google Scholar] [CrossRef]
- Simmonds, N.W. Selection for local adaptation in a plant breeding programme. Theor. Appl. Genet. 1991, 82, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Matsuoka, M. Genetic approaches to crop improvement: Responding to environmental and population changes. Nat. Rev. Genet. 2008, 9, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.J.; Li, Z.J.; Yu, W.W.; Dai, W.S.; Fu, Q.G. Distribution and ecological characteristics of Torreya grandis in China. J. Zhejiang For. Coll. 2007, 24, 383–388. [Google Scholar]
- Kang, N.; Tang, Z.X. Studies on the taxonomy of the genus Torreya. Bull. Bot. Res. 1995, 15, 349–362. [Google Scholar]
- Qiu, Y.X.; Fu, C.X.; Comes, H.P. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 2011, 59, 225–244. [Google Scholar] [CrossRef]
- Liu, J.Q.; Sun, Y.S.; Ge, X.J.; Gao, L.M.; Qiu, Y.X. Phylogeographic studies of plants in China: Advances in the past and directions in the future. J. Syst. Evol. 2012, 50, 267–275. [Google Scholar] [CrossRef]
- Li, Z.J.; Luo, C.F.; Cheng, X.J.; Feng, X.J.; Yu, W.W. Component analysis and nutrition evaluation of seeds of Torreya grandis ‘Merrillii’. J. Zhejiang For. Coll. 2005, 22, 540–544. [Google Scholar]
- Zhang, J.; Hu, L.; Guo, L.; Ren, W.; Zhao, L.; Wang, N.; Zhang, E.; Tang, J.; Chen, X. The maintenance of stable yield and high genetic diversity in the agricultural heritage torreya tree system. BMC Ecol. 2019, 19, 41. [Google Scholar] [CrossRef]
- Li, Z.J.; Cheng, X.J.; Dai, W.S.; Jing, B.H.; Wang, A.G. History and status and development of Torreya grandis in Zhejiang Province. J. Zhejiang For. Coll. 2004, 21, 471–474. [Google Scholar]
- Zeng, J.; Chen, J.; Kou, Y.; Wang, Y. Application of EST-SSR markers developed from the transcriptome of Torreya grandis (Taxaceae), a threatened nut-yielding conifer tree. PeerJ 2018, 6, e5606. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Chen, L.G.; Tong, P.Z. Genetic variation and fingerprinting of Torreya grandis cultivars detected by ISSR markers. Acta Hortic. Sin. 2008, 35, 1125–1130. [Google Scholar] [CrossRef]
- Min, H.; Cheng, S.M.; Kang, Z.X.; Huang, J.Q. Genetic diversity of different Torreya grandis populations by AFLP. For. Res. 2009, 22, 367–372. [Google Scholar]
- Yi, G.M.; Qiu, Y.J. Genetic diversity of Torreya grandis populations as revealed by chloroplast microsatellite markers. J. Fruit Sci. 2014, 31, 583–588. [Google Scholar]
- Chen, T.; Li, J.; Zhang, C.; Gao, Z.; Jin, H.; Dai, W.; Tang, H.; Zhang, M. Genetic structure and diversity of wild populations in Torreya grandis determined with EST-SSR. J. Anhui Agric. Univ. 2020, 47, 224–231. [Google Scholar] [CrossRef]
- Kou, Y.X.; Xiao, K.; Lai, X.R.; Wang, Y.J.; Zhang, Z.Y. Natural hybridization between Torreya jackii and T. grandis (Taxaceae) in southeast China. J. Syst. Evol. 2017, 55, 25–33. [Google Scholar] [CrossRef]
- Wang, Y.J.; Xiao, K.; Kou, Y.X. Torreya jackii (Taxaceae): A special species that is genetically admixed, morphologically distinct, and geographically sympatric with parent species. Forests 2019, 10, 174. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. (2011) Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.A. Detecting population expansion and decline using microsatellites. Genetics 1999, 153, 2013–2029. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar] [CrossRef]
- Kou, Y.; Zhang, L.; Fan, D.; Cheng, S.; Li, D.; Hodel, R.G.J.; Zhang, Z. Evolutionary history of a relict conifer, Pseudotaxus chienii (Taxaceae), in south-east China during the late Neogene: Old lineage, young populations. Ann. Bot. 2020, 125, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Fan, D.; Cheng, S.; Yang, Y.; Wang, M.; Wang, Y.; Zhang, Z. Peripatric speciation within Torreya fargesii (Taxaceae) in the Hengduan Mountains inferred from multi-loci phylogeography. BMC Ecol. Evol. 2023, 23, 74. [Google Scholar] [CrossRef] [PubMed]
- Small, R.L.; Ryburn, J.A.; Cronn, R.C.; Seelanan, T.; Wendel, J.F. The tortoise and the hare: Choosing between noncoding plastome and nuclear Adh sequences for phylogeny reconstruction in a recently diverged plant group. Am. J. Bot. 1998, 85, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, H.L. The hows and whys of cytoplasmic inheritance in seed plants. Am. J. Bot. 1996, 83, 383–404. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Yeh, F.C.; Yang, R.C. POPGENE VERSION 1.31, Microsoft Window-Based Freeware for Population Genetic Analysis. University of Alberta and Tim Boyle, Centre for International Forestry Research. 1999. Available online: https://sites.ualberta.ca/~fyeh/popgene (accessed on 11 July 2023).
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Leblois, R.; Marin, J.M.; Estoup, A. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Möller, M.; Provan, J.; Gao, L.M.; Poudel, R.C.; Li, D.Z. Geological and ecological factors drive cryptic speciation of yews in a biodiversity hotspot. New Phytol. 2013, 199, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Fielding, A.H.; Bell, J.F. A review of methods for the assessment of prediction errors in conservation presence/absence models. Environ. Conserv. 1997, 24, 38–49. [Google Scholar] [CrossRef]
- Fick, S.E.; Hijmans, R.J. WorldClim 2: New 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 2017, 37, 4302–4315. [Google Scholar] [CrossRef]
- Dias, A.; Giovannelli, G.; Fady, B.; Spanu, I.; Vendramin, G.G.; Bagnoli, F.; Carvalho, A.; Silva, M.E.; Lima-Brito, J.; Lousada, J.L.; et al. Portuguese Pinus nigra J.F. Arnold populations: Genetic diversity, structure and relationships inferred by SSR markers. Ann. For. Sci. 2020, 77, 64. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New Forest. 1992, 6, 95–124. [Google Scholar] [CrossRef]
- Prunier, J.; Verta, J.P.; MacKay, J.J. Conifer genomics and adaptation: At the crossroads of genetic diversity and genome function. New Phytol. 2016, 209, 44–62. [Google Scholar] [CrossRef]
- Ge, X.J.; Zhou, X.L.; Li, Z.C.; Hsu, T.W.; Schaal, B.A.; Chiang, T.Y. Low genetic diversity and significant population structuring in the relict Amentotaxus argotaenia complex (Taxaceae) based on ISSR fingerprinting. J. Plant Res. 2005, 118, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; López-Pujol, J.; Wang, H.W.; Ge, S.; Zhang, Z.Y. Molecular evidence for glacial expansion and interglacial retreat during Quaternary climatic changes in a montane temperate pine (Pinus kwangtungensis Chun ex Tsiang) in southern China. Plant Syst. Evol. 2010, 284, 219–229. [Google Scholar] [CrossRef]
- Cun, Y.Z.; Wang, X.Q. Phylogeography and evolution of three closely related species of Tsuga (hemlock) from subtropical eastern Asia: Further insights into speciation of conifers. J. Biogeogr. 2015, 42, 315–327. [Google Scholar] [CrossRef]
- Buchholz, J.T. The embryogeny of Torreya, with a note on Austrotaxus. Bull. Torrey Bot. Club 1940, 67, 731–754. [Google Scholar] [CrossRef]
- Zachos, J.; Pagani, M.; Sloan, L.; Thomas, E.; Billups, K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Location | Latitude (°N) | Longitude (°E) | Ns | cpDNA | nSSR | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nh | Hd | π | θW | HO | HE | I | PIC | |||||
| T. grandis | ||||||||||||
| 1 | Xinning, Hunan, China | 26.57 | 111.16 | 15 | 2 | 0.476 | 0.00019 | 0.00012 | 0.5037 | 0.4279 | 0.7041 | 0.3629 |
| 2 | Ningxiang, Hunan, China | 28.24 | 112.02 | 8 | 1 | 0 | 0 | 0 | 0.5341 | 0.3894 | 0.5682 | 0.3020 |
| 3 | Tonggu, Jiangxi, China | 28.70 | 114.19 | 8 | 2 | 0.536 | 0.00021 | 0.00015 | 0.4481 | 0.4432 | 0.6711 | 0.3509 |
| 4 | Jianou, Fujian, China | 26.89 | 118.65 | 13 | 1 | 0 | 0 | 0 | 0.4326 | 0.3511 | 0.5805 | 0.2954 |
| 5 | Lichuan, Jiangxi, China | 27.04 | 116.92 | 15 | 1 | 0 | 0 | 0 | 0.4851 | 0.4072 | 0.6899 | 0.3504 |
| 6 | Yanshan, Jiangxi, China | 27.88 | 117.75 | 18 | 1 | 0 | 0 | 0 | 0.4778 | 0.4267 | 0.7014 | 0.3610 |
| 7 | Guangfeng, Jiangxi, China | 28.32 | 118.42 | 8 | 1 | 0 | 0 | 0 | 0.4594 | 0.3586 | 0.5364 | 0.2800 |
| 8 | Longquan, Zhejiang, China | 27.95 | 119.25 | 9 | 1 | 0 | 0 | 0 | 0.5000 | 0.3922 | 0.6366 | 0.3251 |
| 9 | Songyang, Zhejiang, China | 28.36 | 119.32 | 14 | 1 | 0 | 0 | 0 | 0.4912 | 0.3788 | 0.6323 | 0.3218 |
| 10 | Jiande, Zhejiang, China | 29.46 | 119.67 | 12 | 1 | 0 | 0 | 0 | 0.4924 | 0.4032 | 0.6909 | 0.3477 |
| 11 | Jingdezhen, Jiangxi, China | 29.54 | 117.66 | 18 | 1 | 0 | 0 | 0 | 0.6035 | 0.4407 | 0.7115 | 0.3658 |
| 12 | Huangshan, Anhui, China | 30.01 | 118.09 | 15 | 1 | 0 | 0 | 0 | 0.4996 | 0.4337 | 0.7203 | 0.3687 |
| 13 | Lin’an, Zhejiang, China | 30.32 | 119.44 | 11 | 2 | 0.327 | 0.00013 | 0.00014 | 0.5159 | 0.4430 | 0.7074 | 0.3667 |
| 14 | Pan’an, Zhejiang, China | 28.98 | 120.50 | 12 | 1 | 0 | 0 | 0 | 0.5241 | 0.4366 | 0.7070 | 0.3619 |
| 15 | Dongyang, Zhejiang, China | 29.45 | 120.47 | 10 | 2 | 0.467 | 0.00206 | 0.00156 | 0.8172 | 0.5048 | 0.7291 | 0.3830 |
| 16 | Zhuji, Zhejiang, China | 29.70 | 120.52 | 17 | 2 | 0.309 | 0.00136 | 0.00130 | 0.8095 | 0.5009 | 0.7352 | 0.3885 |
| T. nucifera | ||||||||||||
| 17 | Nagano, Japan | 35.68 | 137.62 | 6 | 2 | 0.333 | 0.00013 | 0.00017 | 0.1364 | 0.1088 | 0.1469 | 0.0783 |
| T. jackii | ||||||||||||
| 18 | Xianju, Zhejiang, China | 28.58 | 120.59 | 8 | 2 | 0.250 | 0.00010 | 0.00015 | 0.2354 | 0.2140 | 0.3366 | 0.1727 |
| Population | Locus | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| gr12 | gr16 | gr28 | gr29 | gr34 | gr44 | gr48 | gr67 | gr80 | gr81 | gr98 | |
| 1 | 0.04 | 0.19 | 0.19 | 0.60 | - | 0.60 | 15.67 * | 5.08 | 6.22 | 14.00 ** | 5.83 |
| 2 | 0.43 | 2.88 | - | 3.17 | - | 0.04 | 8.00 * | 0.16 | 0.43 | 8.00 * | 2.88 |
| 3 | 0.99 | 0.04 | 2.78 | 3.32 | - | 0.19 | 5.33 | 1.18 | 0.16 | 8.00 * | 3.50 |
| 4 | 0.74 | - | - | 0.22 | 0.43 | 0.11 | 7.30 | 3.94 | 5.08 | 13.00 ** | 6.10 |
| 5 | 3.84 | 0.32 | 0.36 | 3.24 | - | 0.08 | 14.36 * | 0.39 | 0.22 | 13.00 ** | 5.43 |
| 6 | 3.79 | 0.57 | 0.47 | 3.31 | 0.02 | 4.04 | 11.08 * | 9.69 * | 0.02 | 16.00 ** | 2.44 |
| 7 | 0.43 | 0.58 | 0.89 | 0.43 | 0.04 | - | 8.00 * | 6.58 | - | 8.00 ** | 3.26 |
| 8 | 1.48 | 0.14 | 0.74 | 0.36 | 0.03 | - | 9.54 | 1.84 | 0.03 | 9.00 ** | 3.06 |
| 9 | 4.32 | 0.08 | 0.02 | 4.33 | - | 1.04 | 14.00 *** | 1.55 | 0.02 | 14.00 ** | 2.82 |
| 10 | 2.45 | 0.10 | 0.25 | 9.65 * | 0.10 | - | 11.49 | 4.09 | - | 19.47 ** | 6.44 |
| 11 | 5.00 | 3.49 | 4.31 | 1.78 | - | 4.50 | 18.33 ** | 0.33 | 3.87 | 18.00 *** | 1.05 |
| 12 | 0.28 | 8.66 ** | 2.69 | 2.16 | 0.02 | 0.02 | 15.00 ** | 1.62 | 1.39 | 15.00 ** | 3.55 |
| 13 | 0.80 | 0.11 | 0.03 | 0.81 | 0.27 | 0.55 | 7.98 * | 4.24 | 0.55 | 11.00 * | 11.90 |
| 14 | 1.66 | 0.11 | 0.10 | 2.35 | 0.25 | 0.48 | 11.00 * | 0.77 | 0.15 | 12.00 ** | 0.80 |
| 15 | 4.02 | 4.44 * | 4.44 * | 6.69 | 6.69 * | 4.44 * | 10.00 * | 10.00 ** | - | 10.00 * | 20.00 ** |
| 16 | 12.46 *** | 8.33 ** | 8.33 ** | 19.13 *** | 8.33 ** | 7.49 | 17.00 ** | 7.49 ** | 0.07 | 17.00 ** | 24.60 *** |
| Parameter | Mean | Median | Mode | 95% CI | |
|---|---|---|---|---|---|
| N1 | 1.38 × 103 | 1.19 × 103 | 0.71 × 103 | 2.35 × 102 | 3.72 × 103 |
| N1b | 6.85 × 105 | 6.81 × 105 | 3.76 × 105 | 1.56 × 105 | 1.18 × 106 |
| NA | 1.70 × 105 | 1.57 × 105 | 0.33 × 105 | 9.92 × 103 | 3.82 × 105 |
| t1 (years) | 1.22 × 105 | 0.96 × 105 | 0.42 × 105 | 0.19 × 105 | 3.83 × 105 |
| t2 (years) | 3.75 × 106 | 3.60 × 106 | 3.18 × 106 | 0.28 × 106 | 7.35 × 106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Y.; Ou, Q.; Huang, X.; Wang, Y.; Kou, Y. Alien Species Introduction and Demographic Changes Contributed to the Population Genetic Structure of the Nut-Yielding Conifer Torreya grandis (Taxaceae). Forests 2024, 15, 1451. https://doi.org/10.3390/f15081451
Tan Y, Ou Q, Huang X, Wang Y, Kou Y. Alien Species Introduction and Demographic Changes Contributed to the Population Genetic Structure of the Nut-Yielding Conifer Torreya grandis (Taxaceae). Forests. 2024; 15(8):1451. https://doi.org/10.3390/f15081451
Chicago/Turabian StyleTan, Yuming, Qian Ou, Xin Huang, Yujin Wang, and Yixuan Kou. 2024. "Alien Species Introduction and Demographic Changes Contributed to the Population Genetic Structure of the Nut-Yielding Conifer Torreya grandis (Taxaceae)" Forests 15, no. 8: 1451. https://doi.org/10.3390/f15081451