
Identification of RNAi-Related Genes and Transcriptome Assembly of Loblolly Pine (Pinus taeda, L.) Seedlings Exposed to Insect-Specific dsRNA

Abstract
:1. Introduction
2. Materials and Methods
2.1. dsRNA Selection and Synthesis
2.2. Plant Material and Sample Preparation
2.3. RNA-Seq, Read Processing, and De Novo Assembly
2.4. Protein Selection and Identification
2.5. Differential Expression Analysis
2.6. Functional Annotation and Pathway Enrichment
2.7. Phylogenetic Comparison
3. Results
3.1. RNA-Seq, Read Processing, and De Novo Assembly
3.2. Protein Identification
3.3. Differential Expression Analysis
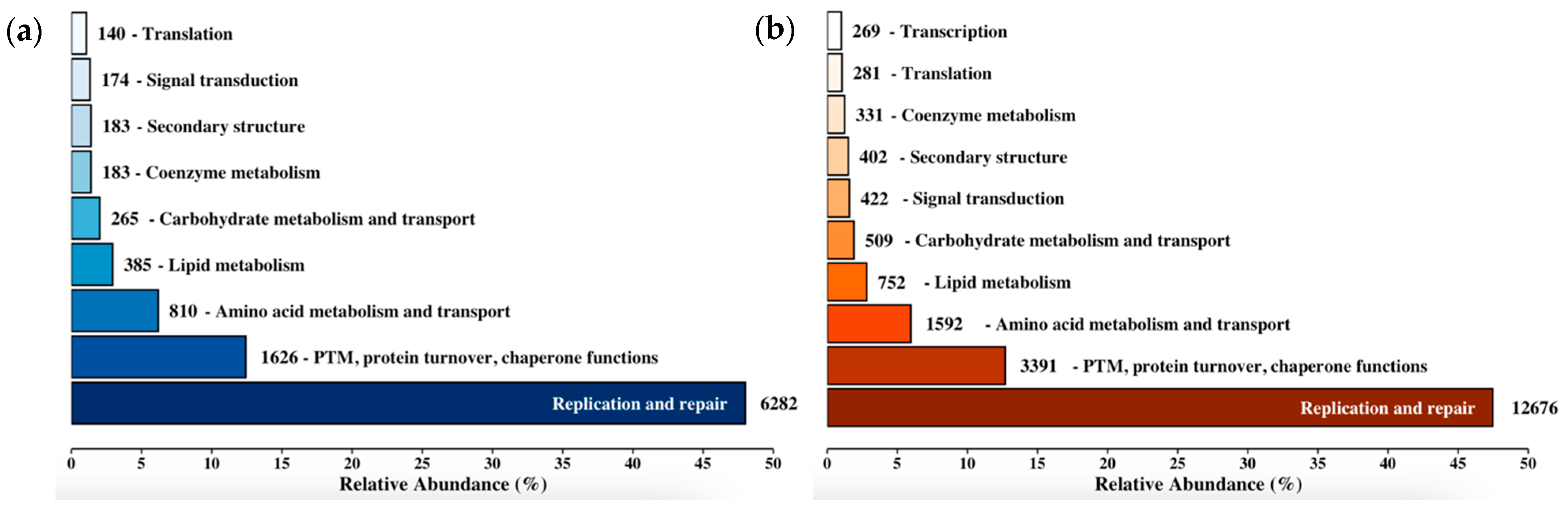
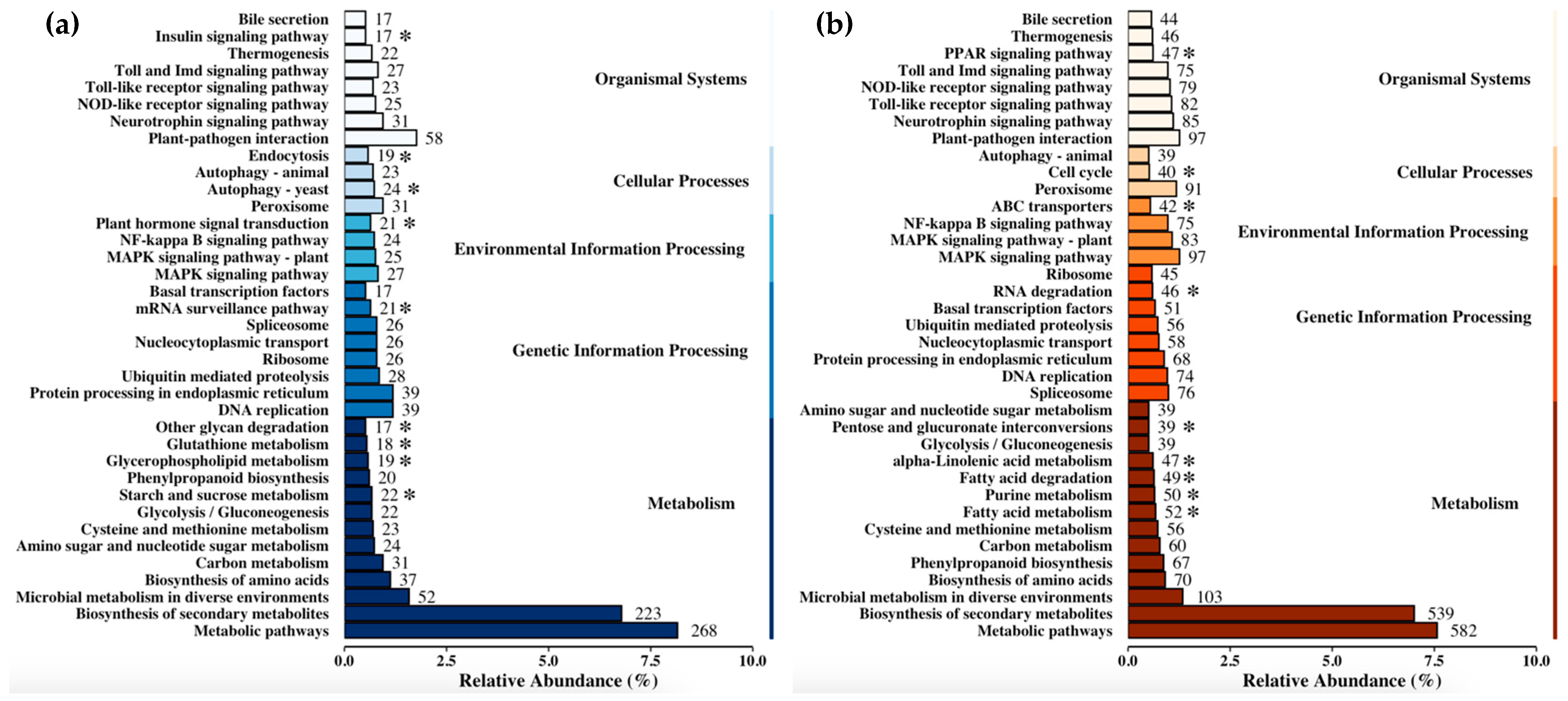
3.4. Functional Annotation and Pathway Enrichment
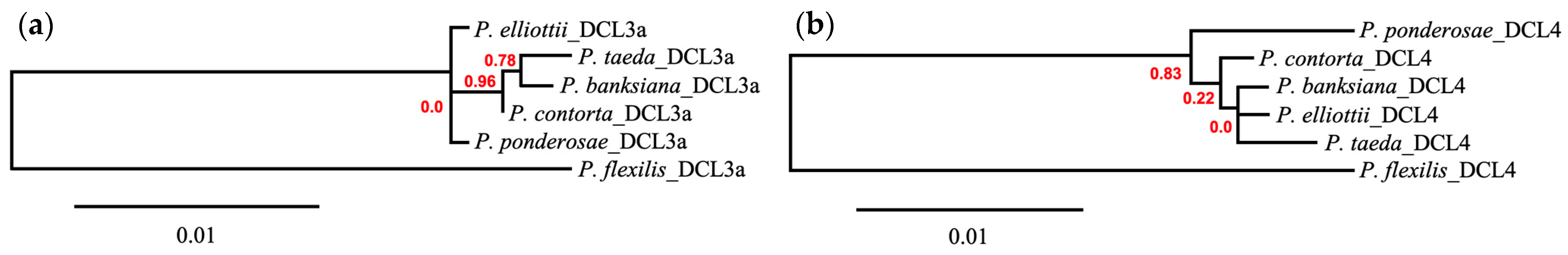
3.5. Phylogenetic Comparison
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Linnakoski, R.; Kasanen, R.; Dounavi, A.; Forbes, K.M. Editorial: Forest Health Under Climate Change: Effects on Tree Resilience, and Pest and Pathogen Dynamics. Front. Plant Sci. 2019, 10, 1157. [Google Scholar] [CrossRef] [PubMed]
- Sicard, P.; Dalstein-Richier, L. Health and vitality assessment of two common pine species in the context of climate change in southern Europe. Environ. Res. 2015, 137, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Han, H.; Chen, S.; Li, M. A fragile soil moisture environment exacerbates the climate change-related impacts on the water use by Mongolian Scots pine (Pinus sylvestris var. mongolica) in northern China: Long-term observations. Agric. Water Manag. 2021, 251, 106857. [Google Scholar] [CrossRef]
- Lin, L.; He, J.; Xie, L.; Cui, G. Prediction of the Suitable Area of the Chinese White Pines (Pinus subsect. Strobus) under Climate Changes and Implications for Their Conservation. Forests 2020, 11, 996. [Google Scholar] [CrossRef]
- Hallingbäck, H.R.; Burton, V.; Vizcaíno-Palomar, N.; Trotter, F.; Liziniewicz, M.; Marchi, M.; Berlin, M.; Ray, D.; Benito Garzón, M. Managing Uncertainty in Scots Pine Range-Wide Adaptation Under Climate Change. Front. Ecol. Evol. 2021, 9, 724051. [Google Scholar] [CrossRef]
- Jactel, H.; Koricheva, J.; Castagneyrol, B. Responses of forest insect pests to climate change: Not so simple. Curr. Opin. Insect Sci. 2019, 35, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Yuan, Y.; Li, X.; Zhang, J. Maximum Entropy Modeling to Predict the Impact of Climate Change on Pine Wilt Disease in China. Front. Plant Sci. 2021, 12, 652500. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gallego, M.; Galiano, L.; Martínez-Vilalta, J.; Stenlid, J.; Capador-Barreto, H.D.; Elfstrand, M.; Camarero, J.J.; Oliva, J. Interaction of drought- and pathogen-induced mortality in Norway spruce and Scots pine. Plant Cell Environ. 2022, 45, 2292–2305. [Google Scholar] [CrossRef] [PubMed]
- Kurz, W.A.; Dymond, C.C.; Stinson, G.; Rampley, G.J.; Neilson, E.T.; Carroll, A.L.; Ebata, T.; Safranyik, L. Mountain pine beetle and forest carbon feedback to climate change. Nature 2008, 452, 987–990. [Google Scholar] [CrossRef]
- Clark, K.L.; Skowronski, N.; Hom, J. Invasive insects impact forest carbon dynamics. Glob. Change Biol. 2010, 16, 88–101. [Google Scholar] [CrossRef]
- Ghimire, B.; Williams, C.A.; Collatz, G.J.; Vanderhoof, M.; Rogan, J.; Kulakowski, D.; Masek, J.G. Large carbon release legacy from bark beetle outbreaks across Western United States. Glob. Change Biol. 2015, 21, 3087–3101. [Google Scholar] [CrossRef]
- Richardson, D.M. Ecology and Biogeography of Pinus; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Johnsen, K.H.; Wear, D.; Oren, R.; Teskey, R.O.; Sanchez, F.; Will, R.; Butnor, J.; Markewitz, D.; Richter, D.; Rials, T.; et al. Meeting Global Policy Commitments: Carbon Sequestration and Southern Pine Forests. J. For. 2001, 99, 14–21. [Google Scholar] [CrossRef]
- Laclau, P. Biomass and carbon sequestration of ponderosa pine plantations and native cypress forests in northwest Patagonia. For. Ecol. Manag. 2003, 180, 317–333. [Google Scholar] [CrossRef]
- Sullivan, T.P.; Sullivan, D.S.; Lindgren, P.M.F.; Ransome, D.B.; Zabek, L. Twenty-Five Years after Stand Thinning and Repeated Fertilization in Lodgepole Pine Forest: Implications for Tree Growth, Stand Structure, and Carbon Sequestration. Forests 2020, 11, 337. [Google Scholar] [CrossRef]
- Raffa, K.F.; Aukema, B.H.; Bentz, B.J.; Carroll, A.L.; Hicke, J.A.; Turner, M.G.; Romme, W.H. Cross-scale Drivers of Natural Disturbances Prone to Anthropogenic Amplification: The Dynamics of Bark Beetle Eruptions. BioScience 2008, 58, 501–517. [Google Scholar] [CrossRef]
- Fettig, C.J.; Egan, J.M.; Delb, H.; Hilszczański, J.; Kautz, M.; Munson, A.S.; Nowak, J.T.; Negrón, J.F. 11—Management tactics to reduce bark beetle impacts in North America and Europe under altered forest and climatic conditions. In Bark Beetle Management, Ecology, and Climate Change; Gandhi, K.J.K., Hofstetter, R.W., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 345–394. [Google Scholar]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.L.; Blau, H.M. A brief history of RNAi: The silence of the genes. FASEB J. 2006, 20, 1293–1299. [Google Scholar] [CrossRef]
- Napoli, C.; Lemieux, C.; Jorgensen, R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. Plant Cell 1990, 2, 279–289. [Google Scholar] [CrossRef]
- Romano, N.; Macino, G. Quelling: Transient inactivation of gene expression in Neurospora crassa by transformation with homologous sequences. Mol. Microbiol. 1992, 6, 3343–3353. [Google Scholar] [CrossRef]
- Cogoni, C.; Macino, G. Post-transcriptional gene silencing across kingdoms. Curr. Opin. Genet. Dev. 2000, 10, 638–643. [Google Scholar] [CrossRef]
- Obbard, D.J.; Gordon, K.H.J.; Buck, A.H.; Jiggins, F.M. The evolution of RNAi as a defence against viruses and transposable elements. Philos. Trans. R. Soc. B Biol. Sci. 2008, 364, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Koonin, E.V. Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. 2008, 23, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Whyard, S.; Singh, A.; Wong, S. Ingested double-stranded RNAs can act as species-specific insecticides. Insect Biochem. Mol. Biol. 2009, 39, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Kyre, B.R.; Rodrigues, T.B.; Rieske, L.K. RNA interference and validation of reference genes for gene expression analyses using qPCR in southern pine beetle, Dendroctonus frontalis. Sci. Rep. 2019, 9, 5640. [Google Scholar] [CrossRef] [PubMed]
- Kyre, B.R.; Bentz, B.J.; Rieske, L.K. Susceptibility of mountain pine beetle (Dendroctonus ponderosae Hopkins) to gene silencing through RNAi provides potential as a novel management tool. For. Ecol. Manag. 2020, 473, 118322. [Google Scholar] [CrossRef]
- Liu, B.; Fu, D.; Ning, H.; Tang, M.; Chen, H. Knockdown of CYP6CR2 and CYP6DE5 reduces tolerance to host plant allelochemicals in the Chinese white pine beetle Dendroctonus armandi. Pestic. Biochem. Physiol. 2022, 187, 105180. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.; Rieske, L.K. Ingestion of Species-Specific dsRNA Alters Gene Expression and Can Cause Mortality in the Forest Pest, Ips calligraphus. Forests 2023, 14, 422. [Google Scholar] [CrossRef]
- Hunter, W.B.; Glick, E.; Paldi, N.; Bextine, B.R. Advances in RNA interference: dsRNA Treatment in Trees and Grapevines for Insect Pest Suppression. Southwest. Entomol. 2012, 37, 85–87. [Google Scholar] [CrossRef]
- Miguel, K.; Scott, J. The next generation of insecticides: dsRNA is stable as a foliar-applied insecticide. Pest Manag. Sci. 2015, 72, 801–809. [Google Scholar] [CrossRef]
- Dalakouras, A.; Jarausch, W.; Buchholz, G.; Bassler, A.; Braun, M.; Manthey, T.; Krczal, G.; Wassenegger, M. Delivery of Hairpin RNAs and Small RNAs Into Woody and Herbaceous Plants by Trunk Injection and Petiole Absorption. Front. Plant Sci. 2018, 9, 1253. [Google Scholar] [CrossRef]
- Ghosh, S.K.B.; Hunter, W.B.; Park, A.L.; Gundersen-Rindal, D.E. Double-stranded RNA Oral Delivery Methods to Induce RNA Interference in Phloem and Plant-sap-feeding Hemipteran Insects. J. Vis. Exp. 2018, 12, e57390. [Google Scholar] [CrossRef] [PubMed]
- Cagliari, D.; Dias, N.P.; Galdeano, D.M.; dos Santos, E.Á.; Smagghe, G.; Zotti, M.J. Management of Pest Insects and Plant Diseases by Non-Transformative RNAi. Front. Plant Sci. 2019, 10, 1319. [Google Scholar] [CrossRef]
- Rodrigues, T.B.; Mishra, S.K.; Sridharan, K.; Barnes, E.R.; Alyokhin, A.; Tuttle, R.; Kokulapalan, W.; Garby, D.; Skizim, N.J.; Tang, Y.-w.; et al. First Sprayable Double-Stranded RNA-Based Biopesticide Product Targets Proteasome Subunit Beta Type-5 in Colorado Potato Beetle (Leptinotarsa decemlineata). Front. Plant Sci. 2021, 12, 728652. [Google Scholar] [CrossRef] [PubMed]
- Bragg, Z.; Rieske, L.K. Spatial Distribution and Retention in Loblolly Pine Seedlings of Exogenous dsRNAs Applied through Roots. Int. J. Mol. Sci. 2022, 23, 9167. [Google Scholar] [CrossRef]
- Kaldis, A.; Berbati, M.; Melita, O.; Reppa, C.; Holeva, M.; Otten, P.; Voloudakis, A. Exogenously applied dsRNA molecules deriving from the Zucchini yellow mosaic virus (ZYMV) genome move systemically and protect cucurbits against ZYMV. Mol. Plant Pathol. 2018, 19, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Biedenkopf, D.; Will, T.; Knauer, T.; Jelonek, L.; Furch, A.C.U.; Busche, T.; Koch, A. Systemic spreading of exogenous applied RNA biopesticides in the crop plant Hordeum vulgare. ExRNA 2020, 2, 12. [Google Scholar] [CrossRef]
- Kiselev, K.V.; Suprun, A.R.; Aleynova, O.A.; Ogneva, Z.V.; Dubrovina, A.S. Physiological Conditions and dsRNA Application Approaches for Exogenously induced RNA Interference in Arabidopsis thaliana. Plants 2021, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.T.L.; Fletcher, S.J.; Brosnan, C.A.; Ghodke, A.B.; Manzie, N.; Mitter, N. RNAi as a Foliar Spray: Efficiency and Challenges to Field Applications. Int. J. Mol. Sci. 2022, 23, 6639. [Google Scholar] [CrossRef]
- Pampolini, F.; Rodrigues, T.B.; Leelesh, R.S.; Kawashima, T.; Rieske, L.K. Confocal microscopy provides visual evidence and confirms the feasibility of dsRNA delivery to emerald ash borer through plant tissues. J. Pest Sci. 2020, 93, 1143–1153. [Google Scholar] [CrossRef]
- Pumplin, N.; Sarazin, A.; Jullien, P.E.; Bologna, N.G.; Oberlin, S.; Voinnet, O. DNA Methylation Influences the Expression of DICER-LIKE4 Isoforms, Which Encode Proteins of Alternative Localization and Function. Plant Cell 2016, 28, 2786–2804. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A Simple and Efficient Method for Isolating RNA from Pine Trees. Plant Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Freedman, A.H.; Gaunt, L. TranscriptomeAssemblyTools. GitHub Repository. 2020. Available online: https://github.com/harvardinformatics/TranscriptomeAssemblyTools (accessed on 29 November 2021).
- Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Weinstein, M.; Schuster-Boeckler, B.; Hulselmans, G.; Sclamons. TrimGalore; v0.6.6; GitHub Repository; GitHub: San Francisco, CA, USA, 2020; Available online: https://github.com/FelixKrueger/TrimGalore/releases/tag/0.6.6 (accessed on 29 November 2021).
- Andrews, S.; Mareq; Mahé, F.; Yi, H.; Brokamp, J.; Reimer, N. FastQC, v0.11.19. GitHub Repository. GitHub: San Francisco, CA, USA, 2020. Available online: https://github.com/s-andrews/FastQC/releases/tag/v0.11.9(accessed on 15 November 2021).
- Grabherr, M.; Haas, B.; Yassour, M.; Levin, J.; Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J. TransDecoder, v5.5.0. GitHub Repository. GitHub: San Francisco, CA, USA, 2018. Available online: https://github.com/TransDecoder/TransDecoder/releases/tag/TransDecoder-v5.5.0(accessed on 29 November 2021).
- Bairoch, A.; Apweiler, R. The SWISS-PROT Protein Sequence Database: Its Relevance to Human Molecular Medical Research. J. Mol. Med. 1997, 75, 312–316. Available online: https://pubmed.ncbi.nlm.nih.gov/9181472/ (accessed on 12 June 2023). [PubMed]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.-P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2021, 31, 8–22. [Google Scholar] [CrossRef]
- Zimin, A.V.; Stevens, K.A.; Crepeau, M.W.; Puiu, D.; Wegrzyn, J.L.; Yorke, J.A.; Langley, C.H.; Neale, D.B.; Salzberg, S.L. An improved assembly of the loblolly pine mega-genome using long-read single-molecule sequencing. GigaScience 2017, 6, giw016. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Posit Team. RStudio: Integrated Development Environment for R. 2022. Available online: http://www.posit.co/ (accessed on 20 April 2022).
- R Core Team. R: A Language and Environment for Statistical Computing. 2022. Available online: https://www.R-project.org/ (accessed on 20 April 2022).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A Genomic Perspective on Protein Families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]
- Jin, W.-T.; Gernandt, D.S.; Wehenkel, C.; Xia, X.-M.; Wei, X.-X.; Wang, X.-Q. Phylogenomic and ecological analyses reveal the spatiotemporal evolution of global pines. Proc. Natl. Acad. Sci. USA 2021, 118, e2022302118. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Feng, Y.; Zhu, Z. Dicer-like (DCL) proteins in plants. Funct. Integr. Genom. 2009, 9, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Qi, Y. RNAi in Plants: An Argonaute-Centered View. Plant Cell 2016, 28, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and Assembly of the 22-Gb Loblolly Pine Genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Kendall, T.; Forsythe, E.S.; Dorantes-Acosta, A.; Li, S.; Caballero-Pérez, J.; Chen, X.; Arteaga-Vázquez, M.; Beilstein, M.A.; Mosher, R.A. Ancient Origin and Recent Innovations of RNA Polymerase IV and V. Mol. Biol. Evol. 2015, 32, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.-H.; Liu, C.; Yuan, H.-W.; Li, P.; Li, Y.; Li, W. Identification and expression profiles of sRNAs and their biogenesis and action-related genes in male and female cones of Pinus tabuliformis. BMC Genom. 2015, 16, 693. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mei, J.; Ren, G. Plant microRNAs: Biogenesis, Homeostasis, and Degradation. Front. Plant Sci. 2019, 10, 360. [Google Scholar] [CrossRef] [PubMed]
- Borsani, O.; Zhu, J.; Verslues, P.E.; Sunkar, R.; Zhu, J.-K. Endogenous siRNAs Derived from a Pair of Natural cis-Antisense Transcripts Regulate Salt Tolerance in Arabidopsis. Cell 2005, 123, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- El-Sappah, A.H.; Yan, K.; Huang, Q.; Islam, M.M.; Li, Q.; Wang, Y.; Khan, M.S.; Zhao, X.; Mir, R.R.; Li, J.; et al. Comprehensive Mechanism of Gene Silencing and Its Role in Plant Growth and Development. Front. Plant Sci. 2021, 12, 705249. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M. Biogenesis of trans-acting siRNAs, endogenous secondary siRNAs in plants. Genes Genet. Syst. 2013, 88, 77–84. [Google Scholar] [CrossRef]
- Garbutt, J.S.; Reynolds, S.E. Induction of RNA interference genes by double-stranded RNA; implications for susceptibility to RNA interference. Insect Biochem. Mol. Biol. 2012, 42, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Bragg, Z.; Rieske, L.K. Feasibility of Systemically Applied dsRNAs for Pest-Specific RNAi-Induced Gene Silencing in White Oak. Front. Plant Sci. 2022, 13, 830226. [Google Scholar] [CrossRef]
- Uslu, V.V.; Bassler, A.; Krczal, G.; Wassenegger, M. High-Pressure-Sprayed Double Stranded RNA Does Not Induce RNA Interference of a Reporter Gene. Front. Plant Sci. 2020, 11, 534391. [Google Scholar] [CrossRef] [PubMed]
- Dubrovina, A.S.; Aleynova, O.A.; Kalachev, A.V.; Suprun, A.R.; Ogneva, Z.V.; Kiselev, K.V. Induction of Transgene Suppression in Plants via External Application of Synthetic dsRNA. Int. J. Mol. Sci. 2019, 20, 1585. [Google Scholar] [CrossRef] [PubMed]
- Nityagovsky, N.N.; Kiselev, K.V.; Suprun, A.R.; Dubrovina, A.S. Exogenous dsRNA Induces RNA Interference of a Chalcone Synthase Gene in Arabidopsis thaliana. Int. J. Mol. Sci. 2022, 23, 5325. [Google Scholar] [CrossRef] [PubMed]
- Pampolini, F.; Rieske, L.K. Foliar Application of dsRNA to Induce Gene Silencing in Emerald Ash Borer: Systemic Distribution, Persistence, and Bioactivity. Forests 2023, 14, 1853. [Google Scholar] [CrossRef]
- Kehr, J.; Kragler, F. Long distance RNA movement. New Phytol. 2018, 218, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Mermigka, G.; Verret, F.; Kalantidis, K. RNA silencing movement in plants. J. Integr. Plant Biol. 2016, 58, 328–342. [Google Scholar] [CrossRef]
- Feinberg, E.H.; Hunter, C.P. Transport of dsRNA into Cells by the Transmembrane Protein SID-1. Science 2003, 301, 1545–1547. [Google Scholar] [CrossRef]
- Huvenne, H.; Smagghe, G. Mechanisms of dsRNA uptake in insects and potential of RNAi for pest control: A review. J. Insect Physiol. 2009, 56, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Gao, X.; Xu, J.; Liang, X.; Li, Q.; Yao, J.; Zhu, K.Y. Clathrin-dependent endocytosis plays a predominant role in cellular uptake of double-stranded RNA in the red flour beetle. Insect Biochem. Mol. Biol. 2015, 60, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Wytinck, N.; Sullivan, D.S.; Biggar, K.T.; Crisostomo, L.; Pelka, P.; Belmonte, M.F.; Whyard, S. Clathrin mediated endocytosis is involved in the uptake of exogenous double-stranded RNA in the white mold phytopathogen Sclerotinia sclerotiorum. Sci. Rep. 2020, 10, 12773. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Deikman, J.; Hendrix, B.; Iandolino, A. Barriers to Efficient Foliar Uptake of dsRNA and Molecular Barriers to dsRNA Activity in Plant Cells. Front. Plant Sci. 2020, 11, 816. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Li, X.; Yang, M.; Shao, Q.; Zhao, Y.; Ma, C.; Wang, P. Autophagy: An Intracellular Degradation Pathway Regulating Plant Survival and Stress Response. Front. Plant Sci. 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed]
- Hafrén, A.; Macia, J.-L.; Love, A.J.; Milner, J.J.; Drucker, M.; Hofius, D. Selective autophagy limits cauliflower mosaic virus infection by NBR1-mediated targeting of viral capsid protein and particles. Proc. Natl. Acad. Sci. USA 2017, 114, E2026–E2035. [Google Scholar] [CrossRef]
- Niehl, A.; Wyrsch, I.; Boller, T.; Heinlein, M. Double-stranded RNAs induce a pattern-triggered immune signaling pathway in plants. New Phytol. 2016, 211, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, C.M.J.; Van der Does, D.; Zamioudis, C.; Leon-Reyes, A.; Van Wees, S.C.M. Hormonal Modulation of Plant Immunity. Annu. Rev. Cell Dev. Biol. 2012, 28, 489–521. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, H. mRNA decay in plants: Both quantity and quality matter. Curr. Opin. Plant Biol. 2017, 35, 138–144. [Google Scholar] [CrossRef]
- Liu, L.; Chen, X. RNA quality control as a key to suppressing RNA silencing of endogenous genes in plants. Mol. Plant 2016, 9, 826–836. [Google Scholar] [CrossRef]
- Williams, D.W.; Liebhold, A.M. Climate change and the outbreak ranges of two North American bark beetles. Agric. For. Entomol. 2002, 4, 87–99. [Google Scholar] [CrossRef]
- Biedermann, P.H.W.; Müller, J.; Grégoire, J.-C.; Gruppe, A.; Hagge, J.; Hammerbacher, A.; Hofstetter, R.W.; Kandasamy, D.; Kolarik, M.; Kostovcik, M.; et al. Bark Beetle Population Dynamics in the Anthropocene: Challenges and Solutions. Trends Ecol. Evol. 2019, 34, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Cullingham, C.I.; Cooke, J.E.K.; Dang, S.; Davis, C.S.; Cooke, B.J.; Coltman, D.W. Mountain pine beetle host-range expansion threatens the boreal forest. Mol. Ecol. 2011, 20, 2157–2171. [Google Scholar] [CrossRef] [PubMed]
- Dodds, K.J.; Aoki, C.F.; Arango-Velez, A.; Cancelliere, J.; D’Amato, A.W.; DiGirolomo, M.F.; Rabaglia, R.J. Expansion of Southern Pine Beetle into Northeastern Forests: Management and Impact of a Primary Bark Beetle in a New Region. J. For. 2018, 116, 178–191. [Google Scholar] [CrossRef]
- Sambaraju, K.R.; Carroll, A.L.; Aukema, B.H. Multiyear weather anomalies associated with range shifts by the mountain pine beetle preceding large epidemics. For. Ecol. Manag. 2019, 438, 86–95. [Google Scholar] [CrossRef]
- Aoki, C.F.; Munro, H.L.; Gandhi, K.J.K. 3—Responses and modeling of southern pine beetle and its host pines to climate change. In Bark Beetle Management, Ecology, and Climate Change; Gandhi, K.J.K., Hofstetter, R.W., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 55–85. [Google Scholar]
- Kyre, B.R.; Rieske, L.K. Using RNAi to silence heat shock protein has congeneric effects in North America’s Dendroctonus bark beetles. For. Ecol. Manag. 2022, 520, 120367. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Contigs | Longest Isoform | |||||||
|---|---|---|---|---|---|---|---|---|
| Trinity “Genes” | Trinity Transcripts | Contig N50 | Med. Contig Length | Avg. Contig Length | Contig N50 | Med. Contig Length | Avg. Contig Length | |
| P. banksiana | 46,780 | 64,450 | 1991 | 713 | 1169.73 | 1968 | 509 | 1037.57 |
| P. contorta | 73,567 | 101,587 | 1830 | 521 | 996.76 | 1697 | 396 | 838.49 |
| P. elliottii | 48,867 | 74,195 | 1890 | 730 | 1140.97 | 1879 | 507 | 1005.69 |
| P. flexilis | 44,887 | 64,931 | 1895 | 701 | 1132.61 | 1893 | 502 | 1005.87 |
| P. ponderosa | 64,858 | 95,480 | 1810 | 513 | 985.10 | 1728 | 371 | 831.15 |
| P. taeda | 60,196 | 91,648 | 1873 | 660 | 1093.72 | 1852 | 457 | 949.05 |
| Experimental | 120,820 | 161,900 | 1386 | 344 | 726.86 | 1073 | 281 | 594.55 |
| RNAi-Related Protein | Top BLASTx Hit | PANTHER Classification | (Symbol) Accession | |
|---|---|---|---|---|
| Description | Comparison | |||
| ARGONAUTE (AGO) | AGO5 [Pinus tabuliformis] (AJP06229.1) | E = 0.0 Bit = 2336 %ID = 98.76% | Family: Eukaryotic Translation Initiation Factor 2C (PTHR22891) Subfamily: Argonaute-1, Isoform A (PTHR22891:SF174) | (AGO1) OR466146 |
| AGO5 [Pinus tabuliformis] (AJP06231.1) | E = 0.0 Bit = 1984 %ID = 98.87% | Family: Eukaryotic Translation Initiation Factor 2C (PTHR22891) Subfamily: Protein Argonaute 4 (PTHR22891:SF157) | (AGO4) OR466147 | |
| AGO7 [Pinus tabuliformis] (AJP06233.1) | E = 0.0 Bit = 1057 %ID = 99.40% | Family: Eukaryotic Translation Initiation Factor 2C (PTHR22891) Subfamily: Protein Argonaute 7 (PTHR22891:SF9) | (AGO7) OR466148 | |
| AMINOPHOSPHOLIPID ATPase (ALA) | Predicted: Phospholipid-Transporting ATPase 3-like [Nelumbo nucifera] (XP_010255676.1) | E = 0.0 Bit = 2024 %ID = 78.80% | Family: Probable Phospholipid-Transporting ATPase (PTHR24092) Subfamily: Phospholipid-Transporting ATPase Dnf1-Related (PTHR24092:SF180) | (ALA1) OR466149 |
| Probable phospholipid-transporting ATPase 4 [Amborella trichopoda] (XP_006853854.1) | E = 0.0 Bit = 1939 %ID = 73.84% | Family: Probable Phospholipid-Transporting ATPase (PTHR24092) Subfamily: Phospholipid-Transporting ATPase IA (PTHR24092:SF150) | (ALA2) OR466150 | |
| ALTERED MERISTEM PROGRAM 1 (AMP1) | Probable Glutamate Carboxypeptidase LAMP1 [Physcomitrium patens] (XP_024382971.1) | E = 0.0 Bit = 802 %ID = 55.78% | Family: N-Acetylated-Alpha-Linked Acidic Dipeptidase (PTHR10404) | (AMP1) OR466151 |
| DICER-LIKE (DCL) | DCL1 [Pinus tabuliformis] (AJP06281.1) | E = 0.0 Bit = 3779 %ID = 99.51% | Family: Dicer-related (PTHR14950) Subfamily: Endoribonuclease Dicer (PTHR14950:SF37) | (DCL1) OR466152 |
| DCL4, partial [Pinus tabuliformis] (AJP06285.1) | E = 0.0 Bit = 2039 %ID = 98.19% | Family: Dicer-related (PTHR14950) Subfamily: Endoribonuclease Dicer Homolog 2 (PTHR14950:SF70) | (DCL2 *) OR466153 | |
| DICER-LIKE (DCL) | DCL3a [Pinus tabuliformis] (AJP06283.1) | E = 0.0 Bit = 2844 %ID = 97.65% | Family: Dicer-related (PTHR14950) Subfamily: Endoribonuclease Dicer Homolog 3 (PTHR14950:SF46) | (DCL3 *) OR466154 |
| DCL4, partial [Pinus tabuliformis] (AJP06285.1) | E = 0.0 Bit = 1209 %ID = 97.30% | Family: Dicer-related (PTHR14950) Subfamily: Dicer Like Protein 4 (PTHR14950:SF15) | (DCL4 *) OR466155 | |
| DAWDLE (DDL) | FHA domain-containing protein DDL-like isoform X1 [Nelumbo nucifera] (XP_010267721.1) | E = 5 × 10−121 Bit = 368 %ID = 59.05% | Family: Nuclear Inhibitor of Protein Phosphatase-1 (PTHR23308) Subfamily: SMAD Nuclear-Interacting Protein 1 (PTHR23308:SF36) | (FHA1) OR466156 |
| DOUBLE-STRANDED RNA BINDING PROTEIN (DRB) | DRB4 [Pinus tabuliformis] (AJP06287.1) | E = 0.0 Bit = 1104 %ID = 96.73% | Family: (PTHR46031) Subfamily: Double-Stranded RNA-Binding Protein 2 (PTHR46031:SF26) | (DRB2) OR466157 |
| ENHANCER OF HUA1 (HEN1) | HEN1 [Pinus tabuliformis] (AJP06305.1) | E = 0.0 Bit = 1959 %ID = 96.32% | Family: HEN1 (PTHR21404) | (HEN1) OR466158 |
| HEAT SHOCK PROTEIN (HSP90) | heat shock 90 kDa protein [Pinus tabuliformis] (QSD59059.1) | E = 0.0 Bit = 988 %ID = 99.80% | Family: Heat Shock Protein 90 Family Member (PTHR11528) | (HSP90 *) OR466159 |
| HYPONASTIC LEAVES (HYL1) | HYL1 [Pinus tabuliformis] (AJP06308.1) | E = 0.0 Bit = 953 %ID = 96.11% | Family: (PTHR46031) Subfamily: BcpLH Protein (PTHR46031:SF35) | (BcpLH) OR466160 |
| RNA-DEPENDENT RNA POLYMERASE (RdRp) | RDR6 [Pinus tabuliformis] (AJP06347.1) | E = 0.0 Bit = 2014 %ID = 80.90% | Family: RNA-Dependent RNA Polymerase (PTHR23079) | (RdRp) OR466161 |
| SILENCING DEFECTIVE 3 (SDE3) | Probable RNA helicase SDE3 [Physcomitrium patens] (XP_024390118.1) | E = 2 × 10−85 Bit = 302 %ID = 34.67% | Family: Cancer/Testis Antigen 55 (PTHR45418) Subfamily: Cancer/Testis Antigen 55 (PTHR45418:SF1) | (SDE3) OR466162 |
| SOMATIC EMBRYOGENESIS RECEPTOR KINASE (SERK) | somatic embryogenesis receptor-like kinase [Larix decidua] (AEF56567.2) | E = 0.0 Bit = 1270 %ID = 98.57% | Family: Somatic Embryogenesis Receptor Kinase 1 (PTHR47988) Subfamily: Somatic Embryogenesis Receptor Kinase 2 (PTHR47988:SF32) | (SERK) OR466163 |
| SERRATE (SE) | serrate RNA effector molecule-like isoform X1 [Macadamia integrifolia] (XP_042479977.1) | E = 0.0 Bit = 816 %ID = 60.41% | Family: Arsenite-Resistance Protein 2 (PTHR13165) | (SE) OR466164 |
| SUPPRESSOR OF GENE SILENCING (SGS3) | SGS3 [Pinus tabuliformis] (AJP06351.1) | E = 0.0 Bit = 1583 %ID = 98.33% | Family: Protein Suppressor of Gene Silencing 3 (PTHR46602) | (SGS3) OR466165 |
| Total Transcripts | Direction | DET | longORF | Predicted Peptides | Functional Annotation | Pathway Enrichment |
|---|---|---|---|---|---|---|
| 85,443 | up | 2629 | 53,226 | 36,988 | 13,085 | 3286 |
| down | 4502 | 108,771 | 76,099 | 26,703 | 7691 | |
| combined | 7131 | 161,997 | 113,087 | 39,788 | 10,977 |
| RNAi Related Protein | AA Length | PANTHER Classification | Accession |
|---|---|---|---|
| ENDORIBONUCLEASE DICER HOMOLOG 3A (DCL3a) | P. banksiana: 1874 P contorta: 1874 P. elliottii: 1397 P. flexilis: 1881 P. ponderosae: 1784 P. taeda: 1874 | Family: Dicer-related (PTHR14950) Subfamily: Endoribonuclease Dicer Homolog 3 (PTHR14950:SF46) | P. banksiana: BK063880 P. contorta: BK063886 P. elliottii: BK063892 P. flexilis: BK063898 P. ponderosae: BK063904 P. taeda: BK063910 |
| ENDORIBONUCLEASE DICER HOMOLOG 4 (DCL4) | P. banksiana: 1682 P contorta: 1717 P. elliottii: 1682 P. flexilis: 1688 P. ponderosae: 1413 P. taeda: 1682 | Family: Dicer-related (PTHR14950) Subfamily: Dicer Like Protein 4 (PTHR14950:SF15) | P. banksiana: BK063881 P. contorta: BK063887 P. elliottii: BK063893 P. flexilis: BK063899 P. ponderosae: BK063905 P. taeda: BK063911 |
| ARGONAUTE 1 (AGO1) | P. banksiana: 1149 P contorta: 1152 P. elliottii: 1145 P. flexilis: 1151 P. ponderosae: 1145 P. taeda: 1145 | Family: Eukaryotic Translation Initiation Factor 2C (PTHR22891) Subfamily: Argonaute-1, Isoform A (PTHR22891:SF174) | P. banksiana: BK063876 P. contorta: BK063882 P. elliottii: BK063888 P. flexilis: BK063894 P. ponderosae: BK063900 P. taeda: BK063906 |
| ARGONAUTE 2 (AGO2) | P. banksiana: 1022 P contorta: 1024 P. elliottii: 1062 P. flexilis: 1040 P. ponderosae: 1026 P. taeda: 1064 | Family: Eukaryotic Translation Initiation Factor 2C (PTHR22891) Subfamily: Protein Argonaute 2-Related (PTHR22891:SF4) | P. banksiana: BK063877 P. contorta: BK063883 P. elliottii: BK063889 P. flexilis: BK063895 P. ponderosae: BK063901 P. taeda: BK063907 |
| ARGONAUTE 4 (AGO4) | P. banksiana: 904 * P contorta: 907 * P. elliottii: 935 P. flexilis: 913 P. ponderosae: 973 P. taeda: 925 | Family: Eukaryotic Translation Initiation Factor 2C (PTHR22891) Subfamily: Protein Argonaute 4 (PTHR22891:SF157) | P. banksiana: BK063878 P. contorta: BK063884 P. elliottii: BK063890 P. flexilis: BK063896 P. ponderosae: BK063902 P. taeda: BK063908 |
| ARGONAUTE 7 (AGO7) | P. banksiana: 1127 P contorta: 1127 P. elliottii: 712 * P. flexilis: 264 * P. ponderosae: 1127 P. taeda: 1127 | Family: Eukaryotic Translation Initiation Factor 2C (PTHR22891) Subfamily: Protein Argonaute 7 (PTHR22891:SF9) | P. banksiana: BK063879 P. contorta: BK063885 P. elliottii: BK063891 P. flexilis: BK063897 P. ponderosae: BK063903 P. taeda: BK063909 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bragg, Z.; Rieske, L.K. Identification of RNAi-Related Genes and Transcriptome Assembly of Loblolly Pine (Pinus taeda, L.) Seedlings Exposed to Insect-Specific dsRNA. Forests 2024, 15, 938. https://doi.org/10.3390/f15060938
Bragg Z, Rieske LK. Identification of RNAi-Related Genes and Transcriptome Assembly of Loblolly Pine (Pinus taeda, L.) Seedlings Exposed to Insect-Specific dsRNA. Forests. 2024; 15(6):938. https://doi.org/10.3390/f15060938
Chicago/Turabian StyleBragg, Zachary, and Lynne K. Rieske. 2024. "Identification of RNAi-Related Genes and Transcriptome Assembly of Loblolly Pine (Pinus taeda, L.) Seedlings Exposed to Insect-Specific dsRNA" Forests 15, no. 6: 938. https://doi.org/10.3390/f15060938