Abstract
Phytophthora nicotianae is a global and polyphagous pathogen with a wide host range. P. nicotianae can infect Areca catechu, Durio zibethinus L., Psidium guajava L., Hevea brasiliensis, and other tree species. The pathogen is capable of inducing butt rot and affecting aerial parts, including stems, leaves, and fruits. Compared to other Phytophthora species, P. nicotianae is more adaptable to abiotic stress. In this study, recombinase polymerase amplification (RPA) in combination with the CRISPR/Cas12a system was used for the detection of P. nicotianae, and achieved rapid and efficient detection of P. nicotianae. The assay was highly specific to P. nicotianae. All 4 tested isolates of P. nicotianae yielded positive results, whereas 30 isolates belonging to 17 other Phytophthora species, 8 fungal species, and 4 Bursaphelenchus xylophilus vermicules lacked detection. Under the conditions of 37 °C, after 20 min of RPA reaction and 25 min of Cas12a cleavage, a DNA concentration as low as 10 pg·μL−1 could be detected. In addition, it detected P. nicotianae from artificially inoculated leaves of Fatsia japonica. In this study, a novel method was established for the efficient and accurate detection of P. nicotianae based on the combination of RPA and the CRISPR/Cas12a system.
1. Introduction
Phytophthora is a genus of plant pathogens that have the potential to cause significant damage to both agricultural and natural ecosystems [1]. Since the 2000s, more than 240 species of phytophthora have been described [2]. The expansion of the genus is likely to continue. Certain Phytophthora species hold greater significance than others in terms of their distribution, host range, and impact on forestry productivity. P. nicotianae causes severe damage to a particularly large number of host plants and is widespread, having been isolated from multiple ecological niches on five continents [3]. P. nicotianae, generally considered a polyphagous pathogen, possesses a wide host range of herbaceous and woody plants, and causes crown rots, and may attack aerial parts, including stems, leaves, and fruits [4]. Phytophthora nicotianae-induced wilt disease was recently discovered on three-year-old potted cycads in Sicily, Italy [5]. In China, P. nicotianae can cause leaf rot disease of Fatsia japonica [6]. In India and Southern Asia, P. nicotianae is one of the main pathogens among Phytophthora spp., and its hosts include, besides citrus and Nicotiana tabacum, Areca catechu, black pepper, Solanum melongena L., Cocos nucifera L., Durio zibethinus L., Psidium guajava L., orchids, Catharanthus roseus, Ananas comosus, Rosa rugosa, Hevea brasiliensis, and fragrant herbs [7]. World agriculture and forestry have undergone major changes as a conjunction of several factors, including globalization and the intensification of international trade, new farming practices, and climate change [8]. Such changes have immediate impacts on plant diseases, with the spreading of pathogens and the modification of their niches. In Cuba, it has been reported that P. nicotianae, which is responsible for leaf rot in avocado, has emerged. Moreover, P. nicotianae is considered the sole pathogen affecting this crop [9]. In South Africa, it is gradually replacing P. cinnamomi in eucalyptus plantations, posing a significant threat to the health of these valuable trees [10]. By detecting diseases early, we can effectively prevent and control their spread, ultimately reducing potential losses. Given the wide occurrence of P. nicotianae and its extensive range of hosts, we aim to establish a rapid and efficient detection technique to assist in the early diagnosis of P. nicotianae and development of disease control strategies.
Currently, traditional morphological identification methods are time-consuming, have low sensitivity, require highly skilled technicians, and often produce inaccurate results [11]. Several molecular detection techniques are available for the identification of P. nicotianae, including duplex PCR [12], multiplex PCR [13,14], and real-time PCR [15,16]. However, these techniques have limitations such as a long reaction time and the need for specialized equipment, which restrict their use in field disease diagnosis [17]. To address this issue, researchers have developed and implemented isothermal amplification-based molecular detection techniques, such as loop-mediated nucleic acid isothermal amplification (LAMP) [18], helicase-dependent amplification (HDA) [19], strand displacement amplification (SDA) [20], rolling circle amplification (RCA) [21], and recombinase polymerase amplification (RPA) [22]. These techniques offer the advantages of rapidity, high specificity, and sensitivity as they can amplify nucleic acids under constant temperature conditions. They have a wide range of applications in nucleic acid testing, gene cloning, sequencing, and more.
RPA, a novel technology developed by Piepenburg et al. (2006) [23], offers a promising approach for the rapid amplification of target nucleic acid from minimally processed samples. With its compact and portable instrumentation, simplicity of operation, rapid amplification speed, and isothermal reaction conditions at 37~42 °C, RPA shows potential as a viable alternative to PCR [24]. Over the past decade, RPA has gained significant traction in diverse detection applications, encompassing bacteria, fungi, parasites, viruses, and drug resistance genes.
The CRISPR-Cas system functions as an adaptive immune system in microorganisms, enabling defense against foreign genetic materials. This system operates by utilizing RNA to direct Cas proteinase to target and cleave specific nucleic acid sequences [25]. Since the initial application of restriction endonucleases for the generation of precise DNA fragments in 1971, researchers have employed prokaryotic molecules for gene editing purposes [26].
The initial success has inspired people to continuously explore new targeting and manipulating nucleic acid systems, including Cas9, Cas12, Cas13 homologous genes, etc. Cas12a orthologs, namely As Cas12a from Acidaminococcus spp. and LbCas12a from Lachnospiraceae spp., were the first to demonstrate activity within mammalian cells, and they specifically recognize the PAM sequence 5′-TTTV positioned upstream of the target sequence [27]. In addition, Cas proteins, including Cas12, Cas13, and Cas14, are capable of recognizing and cleaving target DNA, RNA, or both, and then activate Cas proteins to cleave non-target single-stranded DNA (ssDNA) in trans, without distinction. Cas13 can target RNA, and Cas14 can target single-stranded DNA (ssDNA), but neither can target double-stranded DNA (dsDNA), and Cas12 can directly target dsDNA [28,29,30,31,32]. Specifically, Cas12a and CRISPR RNA (crRNA) form a ribonucleoprotein (RNP) that recognizes protospacer adjacent motif (PAM) sites with the “TTTN” motif and binds to dsDNA complementary to crRNA to form a ternary complex. This specific recognition then activates its trans-cleavage ability [33,34]. Under the action of the activated complex, it can non-specifically cleave the labeled single-stranded DNA reporter probe with a fluorophore-quencher pair. One end of the ssDNA probe is labeled with a reporter fluorophore, and the other end is labeled with a quencher. Once the probe is cleaved by the Cas protein, the quencher is removed, allowing the reporter fluorophore to emit light and achieve visual detection [35,36].
In this study, we have established a highly efficient detection system for tobacco blight based on RPA and CRISPR/Cas12a (Figure 1). The results can be obtained quickly and accurately within 45 min. In general, this method has significantly improved the detection speed and convenience compared with the traditional PCR method. It is of great significance for the effective management of plant diseases.
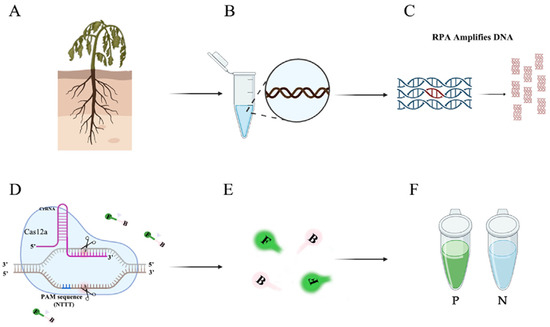
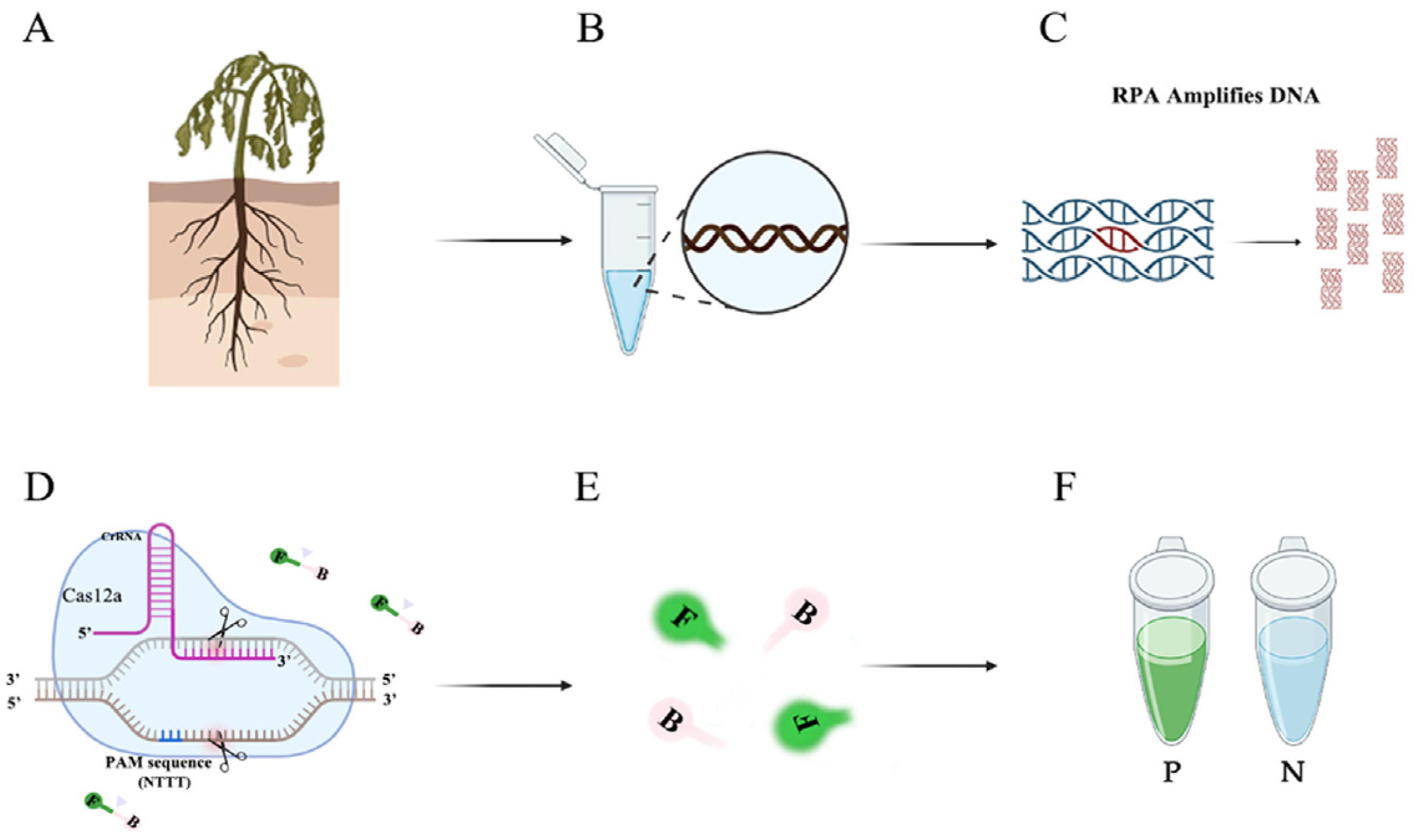
Figure 1.
Flow chart of the RPA-CRISPR/Cas12a assay. (A) Host plants infected with P. nicotianae. (B) Extracting DNA from infected tissues. (C) Amplifying specific primers using RPA technology. (D) CRISPR/Cas12a-based editing. The Cas12a protein can bind to each amplicon and target specific crRNA, activating its cis-cleavage activity. (E) When the single-stranded DNA reporter gene labeled with FAM is cleaved, green fluorescence is visible at an excitation wavelength of 470 nm. (F) P represents a positive result: visible green fluorescence. N represents a negative result: no visible green fluorescence.
2. Materials and Methods
2.1. Maintenance of Isolates and DNA Extraction
The 5 unique P. nicotianae strains, along with the 12 fungi, 33 oomycetes, and 4 nematode isolates from diverse sources detected in this study are detailed in Table 1. These strains were all preserved in the collection of the Department of Plant Pathology at Nanjing Forestry University. Their identities were confirmed through both morphological examination and molecular biology techniques. Under dark conditions at 25 °C, the fungal strains were grown on Potato Dextrose Agar medium (PDA: potato 200 g, dextrose 20 g, agar 20 g, water volume to 1000 mL, and sterilizing at 121 °C for 15 min) for 3–5 days. The oomycete strains were cultured on V8 Juice Agar under dark conditions ranging from 18 to 25 °C (the V8 juice was added with CaCO3 at 1% ratio, and after mixing evenly, the supernatant was obtained by filtering with four layers of gauze. The supernatant was diluted to 10% and then divided into conical bottles, adding 3 g Agar every 200 mL and sterilizing at 121 °C for 15 min.) To propagate, Bursaphelenchus xylophilus were cultivated on the mycelium of Botrytis cinerea for 4–5 days at 25 °C, and allowed to breed [37]. The genomic DNA (gDNA) was extracted using the DNA Secure Plant Kit, which was obtained from Beijing Tian Gen Biotech Co., Ltd. (Beijing, China). The extracted DNA was then quantified using a Nano Drop 1000C spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and appropriately diluted. All DNA samples were stored at −20 °C until further use.

Table 1.
Results for all species used in the specificity test of the RPA-CRISPR/Cas12a assay.
2.2. Design of RPA Primers, crRNA, and ssDNA Reporter
To select the candidate target genes for the P. nicotianae-specific RPA–CRISPR reaction, the annotated genomic sequence of P. nicotianae at NCBI BLAST: Nucleotide Sequence (nih.gov) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (accessed on 9 May 2024) was retrieved. To identify the target genes unique to P. nicotianae, we initially retrieved all the publicly available genome sequences of the Phytophthoa species. Then, all the 26,131 gene sequences of P. nicotianae were used as the queries to search against the above genomes (e-value cutoff: 1 × 10−5), and the genes without any hit were treated as unique to P. nicotianae [38]. The INRA-310 gene was selected as the target for the design of gene-specific RPA primers. We used Primer Premier 6.0 (Premier Biosoft, Palm Alto, CA, USA) software to design RPA primers according to the DNA amplification kit manual. We designed RPA primers targeting the INRA-310 gene (INRA-310 F1: TCTCCAGATGCTTCGTCCGACGGTGAGGATG 277–307 nt; INRA-310 R1: GATCGTACCAACAGTCAAGGAAGACAGGATG 492–522 nt). With a click of a button, the crRNA is designed to target the RPA amplification product of the INRA-310 gene sequence, which lies adjacent to the protospacer adjacent motif (PAM) site of the TTTN spacer region. Precisely aligned with the PAM sequence’s position, we crafted a crRNA that is complementary to the target sequence (crRNA: UAAUUUCUACUAAGUGUAGAUGAGCCCGAGAGUUGUCGAUC) (Figure 2). The single-stranded DNA (ssDNA) reporter program utilized in this study features a 6-FAM label at its 5′ terminus, ensuring robust detection, and is terminated at its 3′ end by the BHQ-1 quencher to prevent unwanted fluorescence. Once the crRNA identifies the target sequence and activates the Cas12a protein, any ssDNA can be cleaved, leading to the activation of the ssDNA reporter gene and the production of a detectable fluorescence. Both the RNA and ssDNA reporter genes were synthesized by GenScript in Nanjing, China and stored at −20 °C ready for further application. The RNA and ssDNA reporter genes, synthesized by GenScript in Nanjing, China, were stored at −20 °C ready for further use.

Figure 2.
Sequence analysis of INRA-310 from Phytophthora nicotianae. Nucleotides targeted by the forward (INRA-310-F1) and reverse (INRA-310-R1) primers; the crRNA sequences are shown below the respective arrows. The arrows indicate the direction of amplification.
2.3. The RPA-CRISPR/Cas12a Assay
The process of employing the RPA-CRISPR/Cas12a method to detect P. nicotianae is diagrammatically represented in Figure 1. This streamlined approach requires a total of 20 min and comprises two distinct steps. In the initial phase, the INRA-310 gene is rapidly amplified in just 15 min using a specific primer pair, INRA-310RPA-F and INRA-310RPA-R, in conjunction with RPA. Subsequently, in the second phase, the CRISPR/Cas12a system is utilized to detect and visualize the presence of the target gene within a mere 5 min.
The RPA reaction was carefully executed in a 50 µL reaction system according to the detailed instructions provided in the kit’s quick guide (Lesunbio, Wuxi, China). Prior to commencing the reaction, each individual mixture was meticulously prepared to include 2 µL of both forward and reverse primers (INRA-310RPA-F/INRA-310RPA-R, 10µM), 25 µL of rehydration buffer provided in the kit, 2 µL of gDNA (100 ng·μL−1), and precisely 16 µL of double distilled water, for a total volume of 47 µL. The reagents were carefully added to the system and briefly centrifuged at 4000 rpm for 5 s to ensure uniform distribution. Subsequently, 3 µL of the promoter was added to the reaction tube cap, which was then securely screwed in place. The mixture was once again centrifuged at 4000 rpm for another 5 s to ensure that the promoter effectively mixed with the premix. After incubating the samples at 37 °C for 5 min, they were gently vortexed 2–3 times, centrifuged at 4000 rpm for another 5 s, and then further incubated at 37 °C for an additional 15 min. To ensure the reliability of the results, each reaction included a no-template control (NTC) and a positive control (PTC). The RPA products of the RPA-CRISPR/Cas12a system were subsequently analyzed by electrophoresis using a 1.5% agarose gel to assess their purity and quantity.
The CRISPR/Cas12a system was conducted in a 50 μL reaction containing various components. These included 38 μL double distilled water, 5 μL 10-fold reaction buffer, 3 μL crRNA (1 μM concentration), 1 μL Cas12a (2 μM concentration) (Magigen, Guangzhou, China), 1 μL single-stranded DNA reporter (10 μM concentration), and 2 μL RPA amplification product. Once the reaction mixture was assembled, it was incubated at 37 °C for 5 min and then observed for fluorescence at a wavelength of 470 nm using a blue LED transmittance instrument (Baisai Ltd., Shanghai, China) or detected using a multi-functional microplate reader (excitation wavelength: 485 nm; emission wavelength: 520 nm). The CRISPR/Cas12a experiments were repeated three times, yielding three results, and standard errors were calculated using STDEVP (numbers 1, 2, and 3). Statistical analysis was performed using GraphPad Prism 8 software (GraphPad Software Inc., San Diego, CA, USA). Differences between the experimental group and the control group were analyzed using the t-test to calculate the p value. p < 0.0001 was considered statistically significant.
2.4. Optimized of RPA-CRISPR/CAS12a for Detection of P. nicotianae
The optimal reaction time of RPA-CRISPR/Cas12a detection of P. nicotianae is divided into two parts: exploring the optimal reaction time of the RPA and CRISPR/Cas12a system. Among them, in the RPA reaction system (as described above) with the mixture added, different reaction times (5 min, 10 min, 15 min, 20 min, 25 min, 30 min, and 35 min) were set at 37 °C under constant temperature conditions. The RPA amplification products were used for CRISPR/Cas12a reactions (consistent with the above reaction system, the reaction time was uniformly set to 30 min) to explore the optimal reaction time of RPA. To eliminate false positives, ddH2O was used instead of gDNA in the RPA system as a negative control (NTC).
Using the RPA amplification products as templates, CRISPR/Cas12a reactions were performed with different concentrations of crRNA (40 nM, 80 nM, 300 nM, 0.5 µM, 0.6 µM, 1 µM, 2 µM, 5 µM, and 10 µM) and single-stranded DNA reporter molecules (40 µM, 500 µM, 1.4 µM, 2 µM, 5 µM, and 10 µM) to determine the optimal detection conditions (Table S1).
2.5. Conventional PCR Assay
A conventional PCR reaction was conducted in a 50 µL reaction mixture containing 25 µL of Prime STAR Max PreMix 2× (Takara Bio, Dalian, China), 21 µL of dd H2O, 2 µL of 100 ng·μL−1 of purified gDNA, and 1 µL of each forward and reverse primer (10 µM). The thermal cycling program began with an initial denaturation step at 94 °C for 3 min. This was followed by 30 s at 94 °C for denaturation, 30 s at 60 °C for annealing, and 45 s at 72 °C for extension, repeated for a total of 33 cycles. The final extension step was performed at 72 °C for 10 min. Amplification was carried out using the Applied Biosystems Veriti Dx 96-Well Thermal Cycler (Thermo Fisher Scientific). Each reaction group included both a positive template control (PTC) and a negative template control (NTC) to ensure accurate results. After amplification, the products were electrophoresed on a 1.5% agarose gel at a voltage of 150 V for approximately 15 min. The gel was then visualized under a UV transilluminator. To ensure reproducibility, the PCR reaction detection was repeated three times.
2.6. Specificity and Sensitivity of RPA-CRISPR/Cas12a Detection Method
A comparative analysis was conducted to assess the specificity and sensitivity of the RPA-CRISPR/Cas12a method for detecting tobacco blight. In this study, pure isolates of gDNA (100 ng·μL−1) were used as templates for both the RPA-CRISPR/Cas12a method and conventional PCR. The strains listed in Table 1 were employed for detection, with positive controls (P. nicotianae isolate, 100 ng·μL−1) and NTCs included in each reaction. This experiment was repeated three times to ensure reproducibility. To evaluate the sensitivity of the method, a series of nine different gDNA dilutions (100 ng·μL−1, 10 ng·μL−1, 1 ng·μL−1, 100 pg·μL−1, 10 pg·μL−1, 1 pg·μL−1, and 100 fg·μL−1) were utilized as templates for both conventional PCR and RPA-CRISPR/Cas12a detection. An NTC was included in each reaction, and each template concentration was tested three times for both methods.
2.7. RPA-CRISPR/Cas12a Method for Detection of P. nicotianae in Artificially Inoculated Fatsia japonica
The detection of Fatsia japonica (Nanjing, China) leaves was carried out using the RPA-CRISPR/Cas12a method. Under dark conditions at 25 °C, P. nicotianae was cultured on V8 agar medium for 3 days. Mycelium blocks were obtained from the edge of the fresh P. nicotianae colony by using a hole punch (diameter 6 mm), and the mycelium was quickly inoculated to the wound site of the treated isolated leaves (stab treatment of the leaves), and allowed to infect and moisturize the leaves. After 3 days of inoculation and culture, obvious disease spots could be seen. After 3 days of inoculation cultivation, DNA was extracted from the leaf sections of both the infected and healthy plants. The RPA-CRISPR/Cas12a detection was carried out according to Figure 1. Pure P. nicotianae gDNA (100 ng·μL−1) and sterile water were used as positive and negative controls, respectively. Positive results showed fluorescence, while negative results did not. The fluorescence was observed at a wavelength of 470 nm using a blue LED transmittance instrument or a multifunctional microplate reader. Each detection method was repeated three times. The CRISPR/Cas12a experiments were repeated three times to obtain three results, and standard errors were calculated using STDEVP. Statistical analysis was performed using IBM SPSS Statistics 23 software. The difference analysis between the experimental group and the control group was carried out using t-tests, and p values were calculated. p < 0.0001 was considered statistically significant in this study (Table S2).
3. Results and Analysis
3.1. Specific Primers for Detecting P. nicotianae
Using primers INRA-310 F1 and INRA-310 R1, a PCR amplification product of around 219 bp was obtained in the conventional PCR reaction (Figure 3). P. hibernalis, P. litorale, P. cinnamomi, P. cryptogea, P. citrophthora, P. capsici, P. palmivora, P. sojae, P. megasperma, P. pini, P. mississippiae, P. ramorum, P. cambivora, P. syringae, Pythium ultimum, Pythium spinosum, Phytopythium helicoides, Pythium aphanidermatum, Botryosphaeria dothidea, Bursaphelenchus xylophilus, Fusarium avenaceum, F. oxysporum, F. asiaticum, F. solaini, F. circinatum, F. proliferatum, and NTC were not detected (Table 1).

Figure 3.
The results of conventional PCR screening for specific target genes. According to the primers designed based on the INRA-310 gene, specific screening was carried out using conventional PCR methods, and detection was performed using 1.5% agarose gel electrophoresis. Only a PCR amplification product of approximately 219 bp was detected in P. nicotianae samples. NTC (negative control, double distilled water). (A) Conventional PCR genus-specific detection results based on the target INRA-310 for P. nicotianae. (B) Conventional PCR species-specific detection results based on the target INRA-310 for P. nicotianae.
3.2. Optimizing the RPA-CRISPR/Cas12a Assay for the Detection of P. nicotianae
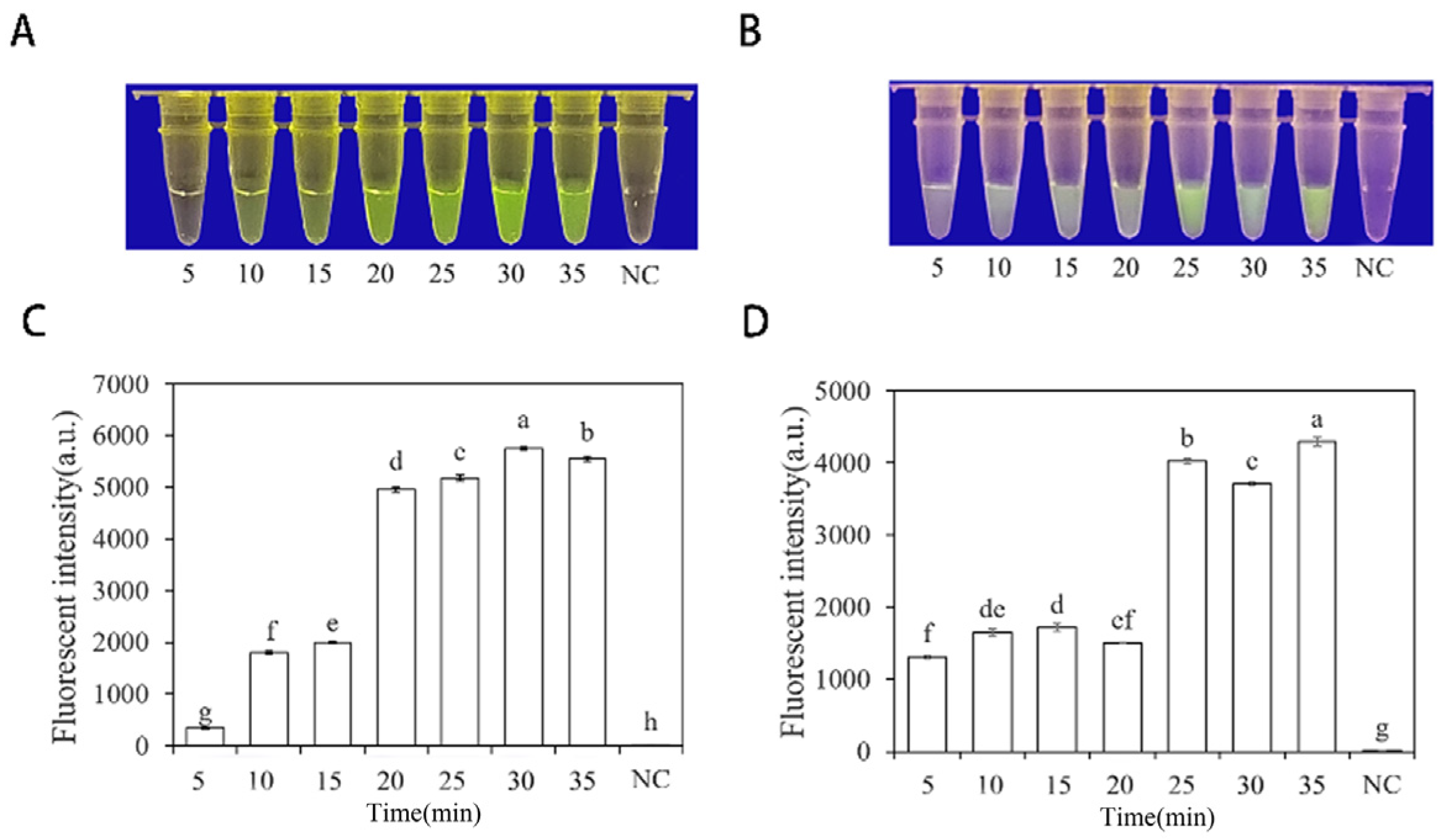
To optimize the concentrations of crRNA probes and ssDNA reporter genes in the RPA-CRISPR/Cas12a assay, different concentrations were tested. The results showed that when the concentrations of crRNA probes and ssDNA reporter genes were 2 μM and 10 μM (Figure 4), respectively, stable results were achieved. To improve experimental efficiency and reduce the required time, different RPA reaction times (5, 10, 15, 20, 25, 30, and 35 min) and different Cas12a cleavage times (5, 10, 15, 20, 25, 30, and 35 min) were tested using a fixed amount of P. nicotianae gDNA template (100 ng·μL−1) for detection. The results showed that fluorescence could be clearly observed after 20 min (Figure 5A,C) of the RPA reaction and after 25 min (Figure 5B,D) of Cas12a cleavage. After evaluating the results, it was determined that the optimal reaction time was 20 min for the RPA reaction and 25 min for Cas12a cleavage. This method enables faster completion of the analysis without compromising its effectiveness.
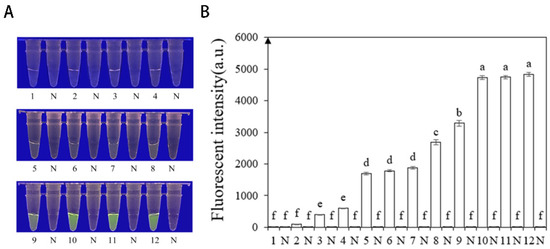
Figure 4.
Optimization of the reaction concentration of CrRNA and ssDNA reporter based on INRA-310 RPA-CRISPR/Cas12a. (A) Observation of green fluorescence at 470 nm using a blue LED transmittance instrument; (B) detection of relative fluorescence intensity using a full-wavelength enzyme microplate reader. 1: 40 nM CrRNA, 40 nM ssDNA reporter. 2: 80 nM CrRNA, 500 nM ssDNA reporter. 3: 300 nM CrRNA, 1.4 nM ssDNA reporter. 4: 0.5 μM CrRNA, 2 μM ssDNA reporter. 5: 0.6 μM CrRNA, 5 μM ssDNA reporter. 6: 1 μM CrRNA, 5 μM ssDNA reporter. 7: 2 μM CrRNA, 5 μM ssDNA reporter. 8: 5 μM CrRNA, 5 μM ssDNA reporter. 9: 1 μM CrRNA, 10 μM ssDNA reporter. 10: 2 μM CrRNA, 10 μM ssDNA reporter. 11: 5 μM CrRNA, 10 μM ssDNA reporter. 12: 10 μM CrRNA, 10 μM ssDNA reporter. N: negative control (ddH2O: no green fluorescence). The one-way ANOVA of the fluorescence readings with those of the negative control showed that p < 0.0001 (different letters).
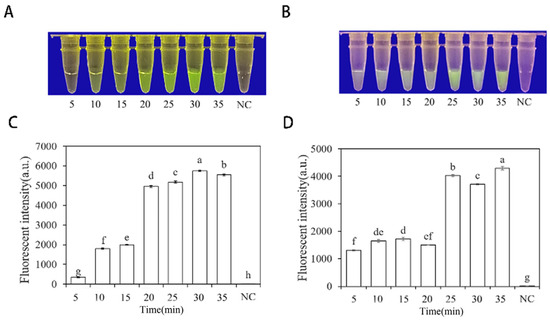
Figure 5.
Optimization of RPA and Cas12a reaction time based on INRA-310-RPA-CRISPR/Cas12a. (A,C) Optimization of RPA reaction time based on INRA-310-RPA-CRISPR/Cas12a. (B,D) Optimization of Cas12a reaction time based on INRA-310-RPA-CRISPR/Cas12a. (A,B) Observation of green fluorescence at 470 nm using a blue LED transmittance instrument; reaction times were 5, 10, 15, 20, 25, 30, and 35 min. (C,D) Detection of relative fluorescence intensity using a full-wavelength enzyme microplate reader; reaction times were 5, 10, 15, 20, 25, 30, and 35 min. NC, negative control (ddH2O: no green fluorescence). The one-way ANOVA of the fluorescence readings with those of the negative control showed that p < 0.0001 (different letters).
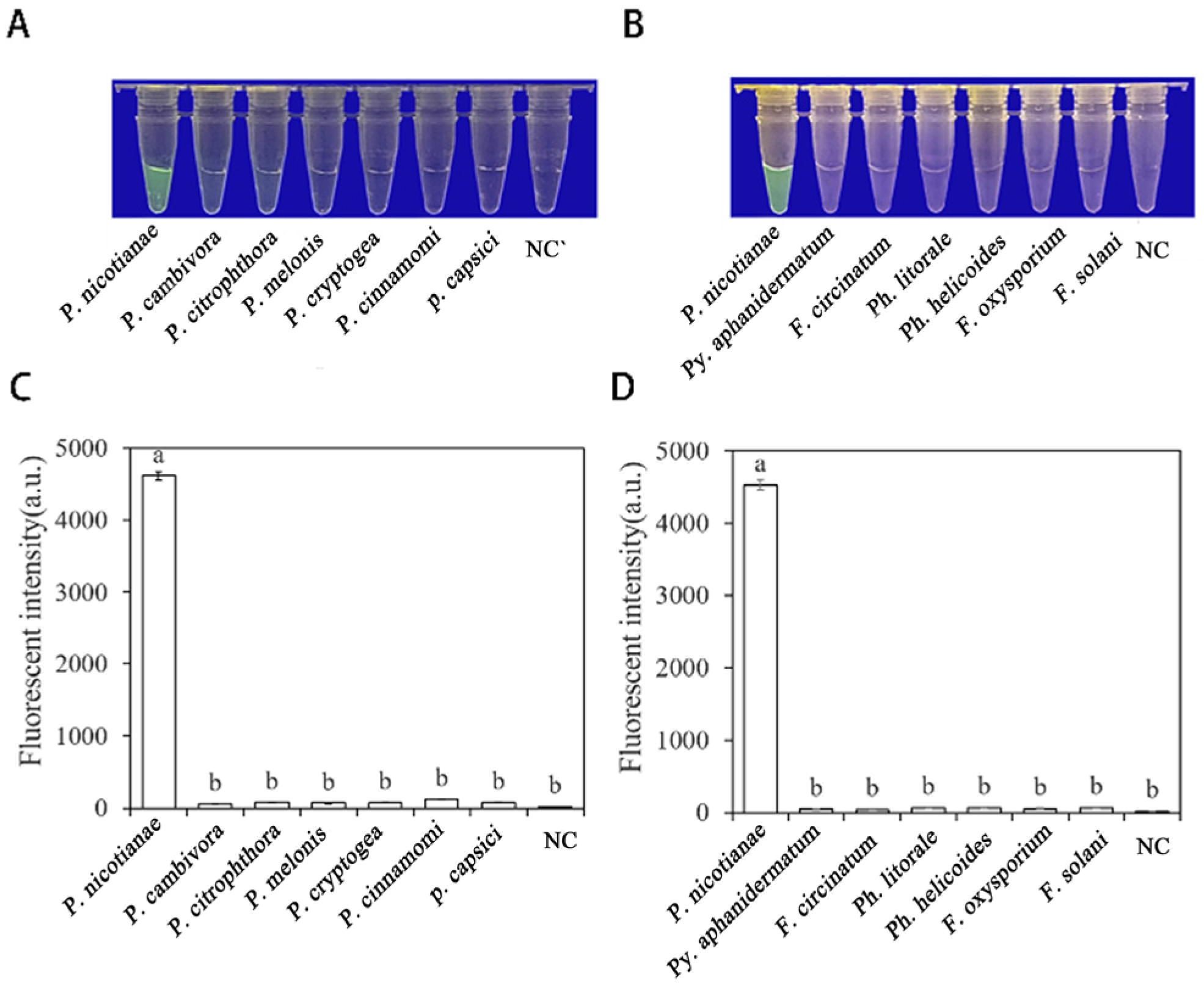
3.3. Specificity of RPA-CRISPR/Cas12a Assay in Rapid Detection of P. nicotianae
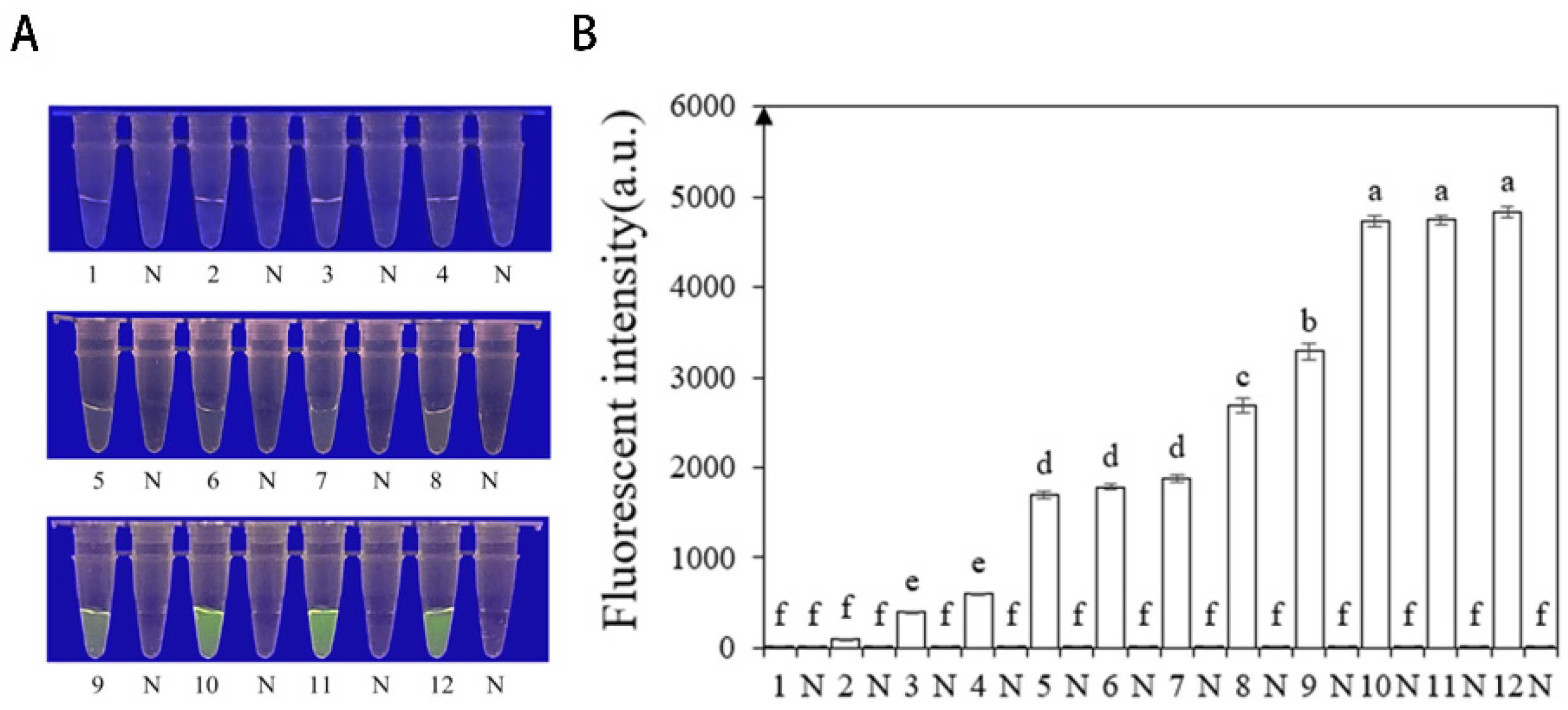
Researchers tested the specificity of their developed RPA-CRISPR/Cas12a method by using DNA of 21 different Phytophthora species, 8 Fungi, and 1 Nematode species (Table 1). The results showed that only when the gDNA of P. nicotianae was used as the RPA reaction template, visible green fluorescence (λex: 485 nm, λem: 520 nm) could be detected using blue LED transilluminators with a wavelength of 470 nm or Aex multifunctional microplate readers. These findings indicate that the RPA-CRISPR/Cas12a detection has high specificity for P. nicotianae (Figure 6).
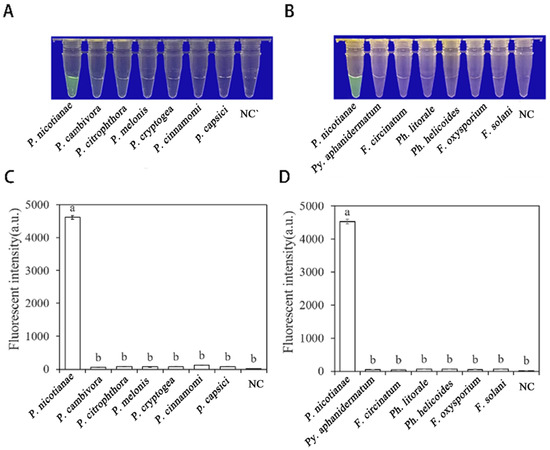
Figure 6.
The specificity of Phytophthora nicotianae was rapidly detected with the RPA- CRISPR/Cas12a assay. (A,C) Evaluation using genomic DNA isolated from P. nicotianae (green fluorescence), P. cambivora (no green fluorescence), P. citrophthora (no green fluorescence), P. melonis (no green fluorescence), P. cryptogea (no green fluorescence), P. cinnamomic (no green fluorescence), P. capsica (no green fluorescence), and negative control (NC; ddH2O: no green fluorescence). (B,D) Evaluation using genomic DNA from P. nicotianae (green fluorescence), Py. Aphanidermatum (no green fluorescence), F. circinatum (no green fluorescence), Ph. Littorale (no green fluorescence), Ph. Helicoides (no green fluorescence), F. oxysporium (no green fluorescence), F. solani (no green fluorescence), and the negative control (NC; ddH2O: no green fluorescence). The one-way ANOVA of the fluorescence readings with those of the negative control showed that p < 0.0001 (different letters).
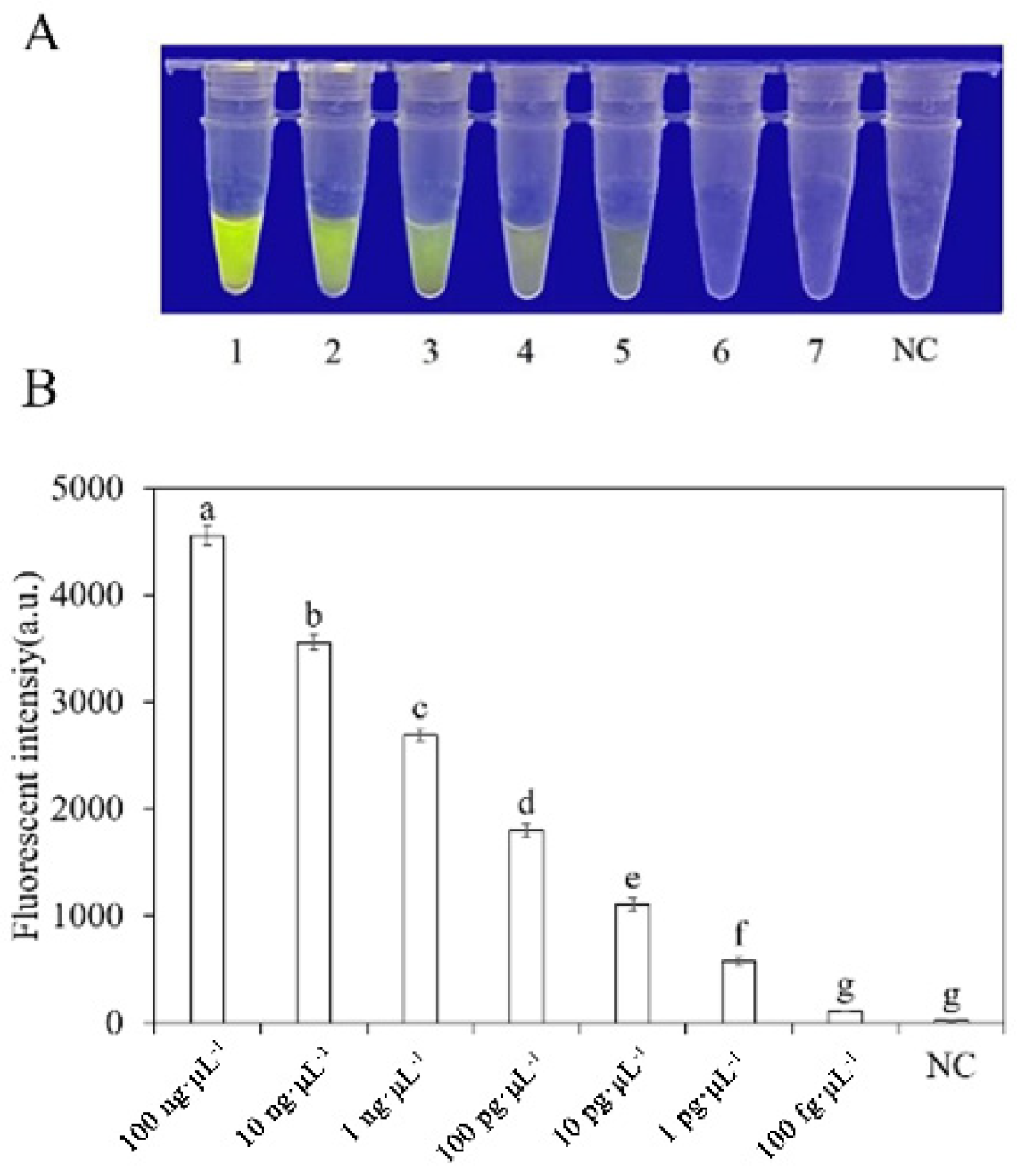
3.4. Determining the Sensitivity of the RPA-CRISPR/Cas12a Assay
To evaluate the sensitivity of the RPA-CRISPR/Cas12a detection of P. nicotianae, we used different concentrations of P. nicotianae gDNA or dd H2O as templates for the RPA reaction. The concentrations used were 100 ng·μL−1, 10 ng·μL−1, 1 ng·μL−1, 100 pg·μL−1, 10 pg·μL−1, 1 pg·μL−1, and 100 fg·μL−1. Subsequently, 2 µL of RPA product was added to the RPA-CRISPR/Cas12a system for analysis. However, when using a blue LED transilluminator with a wavelength of 470 nm, the lowest detection concentration of gDNA was 10 pg·μL−1 (Figure 7).
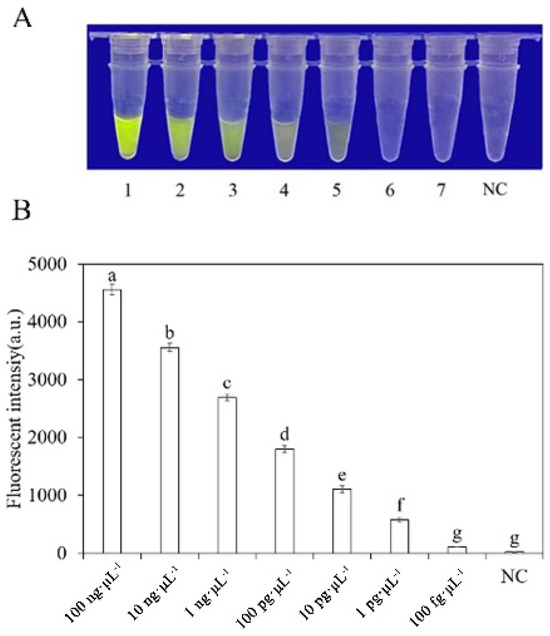
Figure 7.
Analysis of the sensitivity of the RPA-CRISPR/Cas12a detection system. (A) Observation of green fluorescence at 470 nm using a blue LED transmittance instrument; (B) detection of relative fluorescence intensity using a full-wavelength enzyme microplate reader. 1–7: 100 ng·μL−1, 10 ng·μL−1, 1 ng·μL−1, 100 pg·μL−1, 10 pg·μL−1, 1 pg·μL−1, 100 fg·μL−1. N: negative control (ddH2O: no green fluorescence). The one-way ANOVA of the fluorescence readings with those of the negative control showed that p < 0.0001 (different letters).
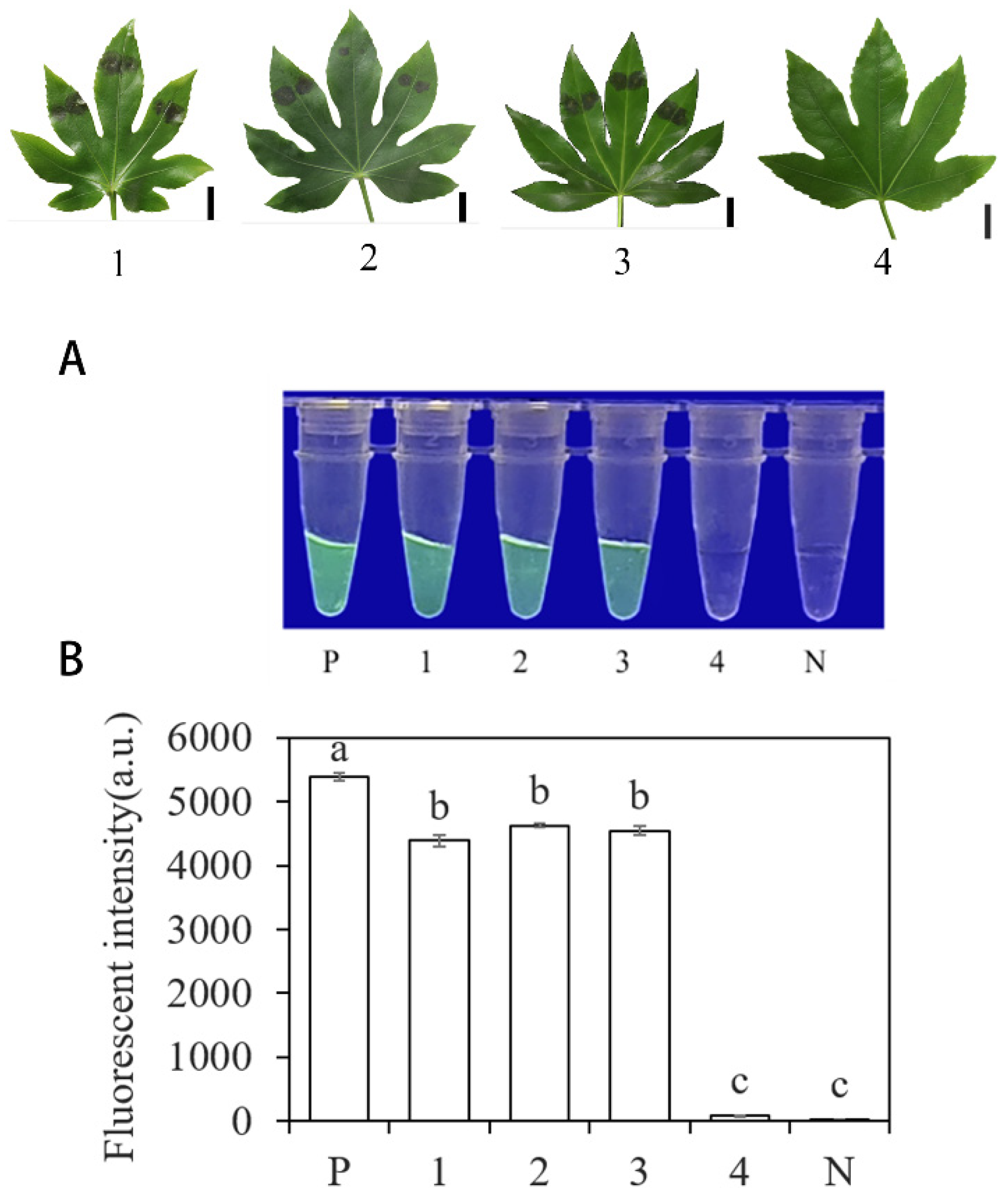
3.5. RPA-CRISPR/Cas12a Method for Detection of P. nicotianae in Artificially Inoculated Fatsia japonica
The plants inoculated with P. nicotianae and healthy plants were subjected to DNA extraction, and the extracted DNA was used as the template for RPA-CRISPR/Cas12a detection. The positive control contained purified gDNA (100 ng·μL−1) of P. nicotianae, and the negative control contained ddH2O. The RPA-CRISPR/Cas12a detection was able to detect the presence of P. nicotianae in the crude DNA samples of inoculated strains and showed green fluorescence. This was observed under a blue LED transmittance instrument at a wavelength of 470 nm and a multifunctional microplate reader with excitation and emission wavelengths of 485 nm and 520 nm, respectively (Figure 8). Controls and NTC samples did not show any fluorescence.
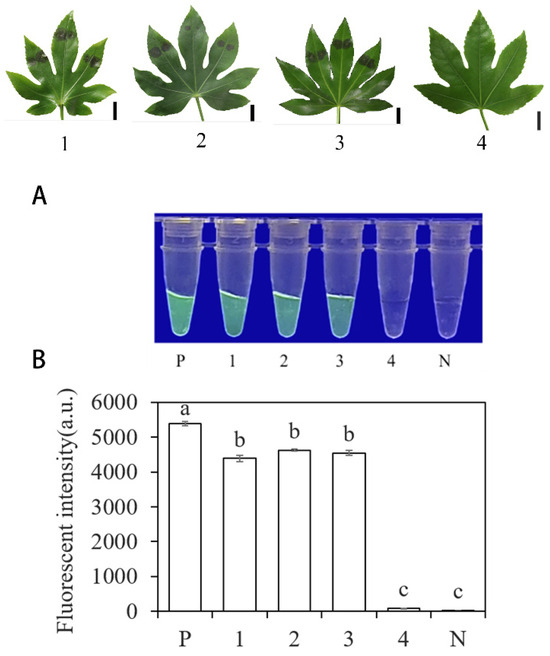
Figure 8.
Detection of artificially inoculated actual samples using the system based on INRA-310-RPA-CRISPR/Cas12a. (A) Observation of green fluorescence at 470 nm using a blue LED transmittance instrument; (B) detection of relative fluorescence intensity using a full-wavelength enzyme microplate reader. P: positive control. 1, 2, 3: artificial inoculation of three strains of plant leaves with Fatsia japonica. 4: leaves of Fatsia japonica plants without inoculation. N: negative control (ddH2O: no green fluorescence). The one-way ANOVA of the fluorescence readings with those of the negative control showed that p < 0.0001 (different letters).
4. Discussion
In this study, we present a method that can detect P. nicotianae in only 45 min, achieving rapid and efficient results through dual recognition technology of RPA and CRISPR-Cas12, and allows observation under UV light and fluorescent dyes. During the experimental process, we made intriguing observations worth highlighting: the outcome is influenced by various factors, with the concentration of crRNA and ssDNA playing a pivotal role in determining its success [39]. Specifically, inadequate concentrations of crRNA and ssDNA can lead to elusive results, while excessively high levels can inflate costs. To identify the most effective concentration blend, we devised twelve distinct combinations and ultimately concluded that a blend of 2 μM crRNA and 10 μM ssDNA maintains result visualization integrity while minimizing reagent wastage.
In addition, under the same conditions, 8 species of fungi, 21 species of oomycetes, and 1 species of nematodes were tested (Table 1). The results showed that only P. nicotianae could be observed under UV light. By testing the diseased parts on the artificially inoculated plants of Fatsia japonica, we can obtain accurate results, which further confirms that this technique can effectively detect P. nicotianae in field environments. This finding is of great significance for the prevention and control of P. nicotianae, as it provides a rapid and accurate detection method that can help to detect and contain the spread of P. nicotianae in a timely manner.
Compared to traditional PCR-based detection methods for diseases, this study offers a more efficient approach that does not require expensive instruments or highly specialized technical skills. While numerous detection methods have been previously explored, including the combination of loop-mediated isothermal amplification (LAMP) with CRISPR/Cas12a to develop a novel diagnostic technique [40], two recently characterized CRISPR nucleases, Cas13 and Cas14, have also been developed for nucleic acid testing [28,41]. However, these methods either require higher isothermal temperatures or involve additional steps to generate target ssDNA substrates from dsDNA amplicons. Additionally, crRNA is prone to degradation by ubiquitous RNases, which can lead to false negatives in field testing [41,42]. Therefore, we believe that the combination of RPA with CRISPR/Cas12a offers a more effective tool for detecting pathogens. Currently, some scholars have used the LAMP method to detect P. nicotianae, but the total time from sample collection to result is around 120 min [18]. In contrast, RPA-CRISPR/Cas12a greatly reduces the detection time.
The present study successfully developed a detection method based on RPA-CRISPR/Cas12a, which enables specific and rapid identification of P. nicotianae. It is noteworthy that this detection method does not require high technical expertise from operators, eliminates the need for expensive instruments and equipment, and maintains a reaction temperature of 37 °C, well within the tolerable range for human handling. These features contribute to enhanced convenience and safety throughout the entire detection process. By employing this efficient and specific detection technology, early monitoring and timely warning of P. nicotianae can be achieved. This approach not only facilitates disease control measures in a timely manner to minimize economic losses but also aids in formulating more scientifically sound strategies for disease prevention and control.
5. Conclusions
In this study, we have established a highly efficient detection method for P. nicotianae based on RPA-CRISPR/Cas12a, which is of great significance for ensuring the healthy development of various industries potentially threatened by P. nicotianae. In addition, the successful establishment of this detection method also provides useful references for the detection of other plant diseases.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/f15060952/s1. Table S1: Screening for crRNA and ssDNA reporter concentrations in the RPA-CRISPR/Cas12a assay system for Phytophthora nicotianae; Table S2: The fluorescence values measured by RPA-CRISPR/Cas12a assay for detection of Phytophthora nicotianae.
Author Contributions
J.Z. (Jiahui Zang) conceptualized and designed the research, analyzed the data, interpreted the results, performed the experiments, and wrote the manuscript. T.D. and T.L. revised the manuscript and directed the project. X.X. and J.Z. (Jing Zhou) analyze the data and make changes to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Key R&D Program of China (2023YFD1401304), Natural Science Foundation of Jiangsu Province (BK20231291), Jiangsu University Natural Science Research Major Project (21KJA220003), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Data Availability Statement
All data generated or analyzed during this study are included in this article.
Acknowledgments
The authors wish to thank Jing Zhou at Nanjing Forestry University for providing Phytophthora nicotianae species isolates that were used in the evaluation of specificity in this study. The authors also thank Danyu Shen for providing support and suggestions during the bioinformatics analyses.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kroon, L.P.; Brouwer, H.; De Cock, A.W.; Govers, F. The genus Phytophthora anno 2012. Phytopathology 2012, 102, 348–364. [Google Scholar] [CrossRef]
- Martin, F.N.; Blair, J.E.; Coffey, M.D. A combined mitochondrial and nuclear multilocus phylogeny of the genus Phytophthora. Fungal Genet. Biol. 2014, 66, 19–32. [Google Scholar] [CrossRef]
- Meng, Y.; Zhang, Q.; Ding, W.; Shan, W. Phytophthora parasitica: A model oomycete plant pathogen. Mycology 2014, 5, 43–51. [Google Scholar] [CrossRef]
- Panabières, F.; Ali, G.S.; Allagui, M.B.; Dalio, R.J.; Gudmestad, N.C.; Kuhn, M.; Roy, S.G.; Schena, L.; Zampounis, A. Phytophthora nicotianae diseases worldwide: New knowledge of a long-recognised pathogen. Phytopathol. Mediterr. 2016, 55, 20–40. [Google Scholar]
- Aloi, F.; Parlascino, R.; Conti, T.S.; Faedda, R.; Pane, A.; Cacciola, S.O. Phytophthora pseudocryptogea, P. nicotianae and P. multivora Associated to Cycas revoluta: First Report Worldwide. Plants 2023, 12, 1197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, T.; Xu, X.; Dai, T.; Liu, T. The New Report of Root Rot on Fatsia japonica Caused by Phytophthora nicotianae in China. Forests 2023, 14, 1459. [Google Scholar] [CrossRef]
- Zhang, K.; Sun, Z.; Shi, K.; Yang, D.; Bian, Z.; Li, Y.; Gou, H.; Jiang, Z.; Yang, N.; Chu, P.; et al. RPA-CRISPR/Cas12a-Based Detection of Haemophilus parasuis. Animals 2023, 13, 3317. [Google Scholar] [CrossRef] [PubMed]
- Guha, R.S.; Grünwald, N. The plant destroyer genus Phytophthora in the 21st century. Rev. Plant Pathol. 2014, 6, 387–412. [Google Scholar]
- Popkin, B. Urbanization, lifestyle changes and the nutrition transition. World Dev. 1999, 27, 1905–1916. [Google Scholar] [CrossRef]
- Machado, M.; Collazo, C.; Pena, M.; Coto, O.; Lopez, M.O. First report of root rot caused by Phytophthora nicotianae in avocado trees (Persea americana) in Cuba. New Dis. Rep. 2013, 28, 9. [Google Scholar] [CrossRef]
- Chen, X.; Ma, K.; Yi, X.; Xiong, L.; Wang, Y.; Li, S. The rapid and visual detection of methicillin-susceptible and methicillin-resistant Staphylococcus aureus using multiplex loop-mediated isothermal amplification linked to a nanoparticle-based lateral flow biosensor. Antimicrob. Resist. Infect. Control. 2020, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Jiang, S.; Feng, S.; Shang, W.; Xing, G.; Qiu, R.; Li, C.; Li, S.; Zheng, W. A Duplex PCR Assay for Rapid Detection of Phytophthora nicotianae and Thielaviopsis basicola. Plant Pathol. J. 2019, 35, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Das, A.K.; Nerkar, S.; Gawande, N.; Thakre, N.; Kumar, A. SCAR marker for Phytophthora nicotianae and a multiplex PCR assay for simultaneous detection of P. nicotianae and Candidatus Liberibacter asiaticus in citrus. J. Appl. Microbiol. 2019, 127, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Asano, T.; Suga, H.; Kageyama, K. A Multiplex PCR for the Detection of Phytophthora nicotianae and P. cactorum, and a Survey of Their Occurrence in Strawberry Production Areas of Japan. Plant Dis. 2011, 95, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Inada, M.; Watanabe, H.; Suga, H.; Kageyama, K. Simultaneous detection and quantification of Phytophthora nicotianae and P. cactorum, and distribution analyses in strawberry greenhouses by duplex real-time PCR. Microbes Environ. 2013, 28, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Blaya, J.; Lacasa, C.; Lacasa, A.; Martínez, V.; Santísima-Trinidad, A.B.; Pascual, J.A.; Ros, M. Characterization of Phytophthora nicotianae isolates in southeast Spain and their detection and quantification through a real-time TaqMan PCR. J. Sci. Food Agric. 2015, 95, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhou, J.; Zheng, Y.; Gamson, A.S.; Roembke, B.T.; Nakayama, S.; Sintim, H.O. Isothermal amplified detection of DNA and RNA. Mol. Biosyst. 2014, 10, 970–1003. [Google Scholar] [CrossRef]
- Hieno, A.; Li, M.; Otsubo, K.; Suga, H.; Kageyama, K. Multiplex LAMP Detection of the Genus Phytophthora and Four Phytophthora Species, P. ramorum, P. lateralis, P. kernoviae, and P. nicotianae, with a Plant Internal Control. Microbes Environ. 2021, 36, ME21019. [Google Scholar] [CrossRef] [PubMed]
- Barreda-García, S.; Miranda-Castro, R.; de-Los-Santos-Álvarez, N.; Miranda-Ordieres, A.J.; Lobo-Castañón, M.J. Helicase-dependent isothermal amplification: A novel tool in the development of molecular-based analytical systems for rapid pathogen detection. Anal. Bioanal. Chem. 2018, 410, 679–693. [Google Scholar] [CrossRef]
- Walker, G.T.; Fraiser, M.S.; Schram, J.L.; Little, M.C.; Nadeau, J.G.; Malinowski, D.P. Strand displacement amplification—An isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 1992, 20, 1691–1696. [Google Scholar] [CrossRef]
- Goo, N.I.; Kim, D.E. Rolling circle amplification as isothermal gene amplification in molecular diagnostics. BioChip J. 2016, 10, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Miles, T.D.; Martin, F.N.; Coffey, M.D. Development of rapid isothermal amplification assays for detection of Phytophthora spp. in plant tissue. Phytopathology 2015, 105, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA Detection using recombination proteins. PloS Biol. 2016, 4, e204. [Google Scholar] [CrossRef]
- Li, J.; Macdonald, J.; von Stetten, F. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 2018, 144, 31–67. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef]
- Danna, K.; Nathans, D. Specific cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae. Proc. Natl. Acad. Sci. USA 1979, 68, 2913–2917. [Google Scholar] [CrossRef] [PubMed]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic Acid Detection With CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA Destruction by Miniature CRISPR-Cas14 Enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, Q.X.; Liu, J.K.; Nie, X.Q.; Zhao, G.P.; Wang, J. CRISPRCas12a has Both Cis- and Trans-Cleavage Activities on Single-Stranded DNA. Cell Res. 2018, 28, 491–493. [Google Scholar] [CrossRef]
- Wang, B.; Wang, R.; Wang, D.; Wu, J.; Li, J.; Wang, J.; Liu, H.; Wang, Y. Cas12aVDet: A CRISPR/Cas12a-Based Platform for Rapid and Visual Nucleic Acid Detection. Anal. Chem. 2019, 91, 12156–12161. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qiu, X.; Xu, S.; Che, Y.; Han, L.; Kang, Y.; Yue, Y.; Chen, S.; Li, F.; Li, Z. A CRISPR-Cas12a-Assisted Fluorescence Platform for Rapid and Accurate Detection of Nocardia cyriacigeorgica. Front. Cell. Infect. Microbiol. 2022, 12, 835213. [Google Scholar] [CrossRef]
- Xiong, Y.; Cao, G.; Chen, X.; Yang, J.; Shi, M.; Wang, Y.; Nie, F.; Huo, D.; Hou, C. One-pot platform for rapid detecting virus utilizing recombinase polymerase amplification and CRISPR/Cas12a. Appl. Microbiol. Biotechnol. 2022, 106, 4607–4616. [Google Scholar] [CrossRef]
- Su, A.; Liu, Y.; Cao, X.; Xu, W.; Liang, C.; Xu, S. A universal CRISPR/Cas12a-mediated AuNPs aggregation-based surface-enhanced Raman scattering (CRISPR/Cas-SERS) platform for virus gene detection. Sens. Actuators B-Chem. 2022, 369, 132295. [Google Scholar] [CrossRef]
- Ramachandran, A.; Huyke, D.A.; Sharma, E.; Huang, M.K.; Banaei, C.N.; Pinsky, B.A.; Santiago, J.G. Electric field-driven microfluidics for rapid CRISPR-based diagnostics and its application to detection of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 29518–29525. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Baker, M.B.; Bao, G.; Searles, C.D. MicroRNA Detection Using a Double Molecular Beacon Approach: Distinguishing Between miRNA and Pre-miRNA. Theranostics 2017, 7, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Hu, J.; Chen, Y.; Tan, J.; Ye, J. Cloning, Bioinformatics Analysis and Physiological Function of the Pine Wood Nematode Bxadh2 Gene. Forests 2023, 14, 1283. [Google Scholar] [CrossRef]
- Tingting, D.; Xiao, Y.; Tao, H. A Novel LAMP Assay for the Detection of Phytophthora cinnamomi Utilizing a New Target Gene Identified From Genome Sequences. Plant Dis. 2019, 103, 3101–3107. [Google Scholar]
- Nagel, J.H.; Gryzenhout, M.; Slippers, B.; Wingfield, M.J. The occurrence and impact of Phytophthora on the african continent. In Phytophthora—A Global Perspective; Lamour, E., Ed.; CAB International: Wallingford, Oxfordshire, UK, 2013; pp. 204–214. [Google Scholar]
- Jiao, J.; Kong, K.; Han, J.; Song, S.; Bai, T.; Song, C.; Wang, M.; Yan, Z.; Zhang, H.; Zhang, R.; et al. Field detection of multiple RNA viruses/viroids in apple using a CRISPR/Cas12a-based visual assay. Plant Biotechnol. 2021, 19, 394–405. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef]
- Shi, Y.; Kang, L.; Mu, R.; Xu, M.; Duan, X.; Li, Y.; Yang, C.; Ding, J.W.; Wang, Q.; Li, S. CRISPR/Cas12a-Enhanced Loop-Mediated Isothermal Amplification for the Visual Detection of Shigella flexneri. Front. Bioeng. Biotechnol. 2022, 10, 845688. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).