
Deciphering the Regulatory Mechanism of PmMYB21 in Early Flowering of Prunus mume through Dap-Seq and WGCNA Analysis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Growth Conditions
2.3. PmMYB21 Full-Length CDs Clone
2.4. Dap-Seq and Data Analysis
2.5. RNA-Seq and WGCNA
2.6. Quantitative Real-Time-PCR Validation
2.7. Candidate Gene Screening
2.8. Heat Map of Candidate Genes
3. Results
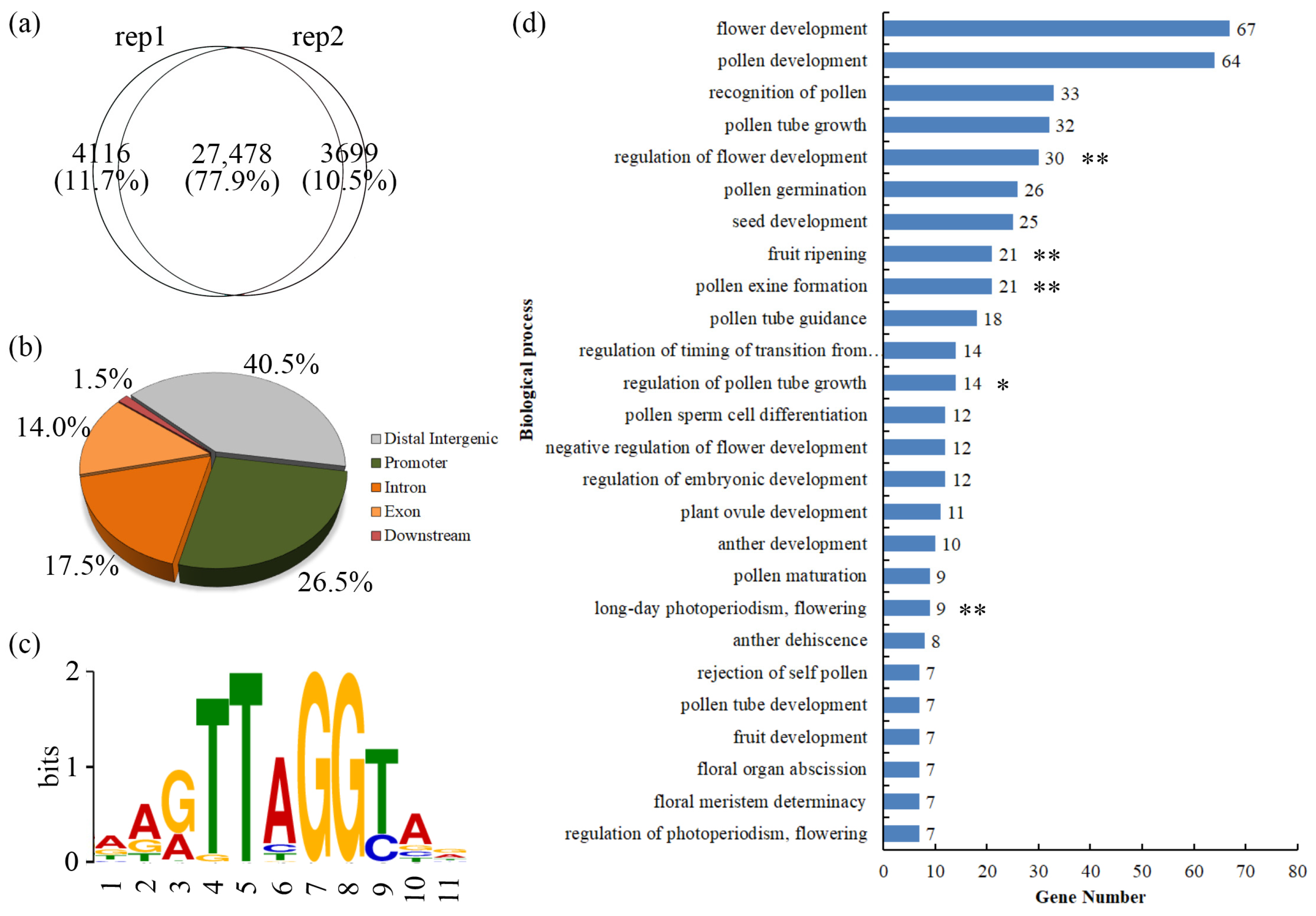
3.1. The Genes Bound by PmMYB21 Are Enriched in the Flowering Process
3.2. The Co-Expression Gene of PmMYB21
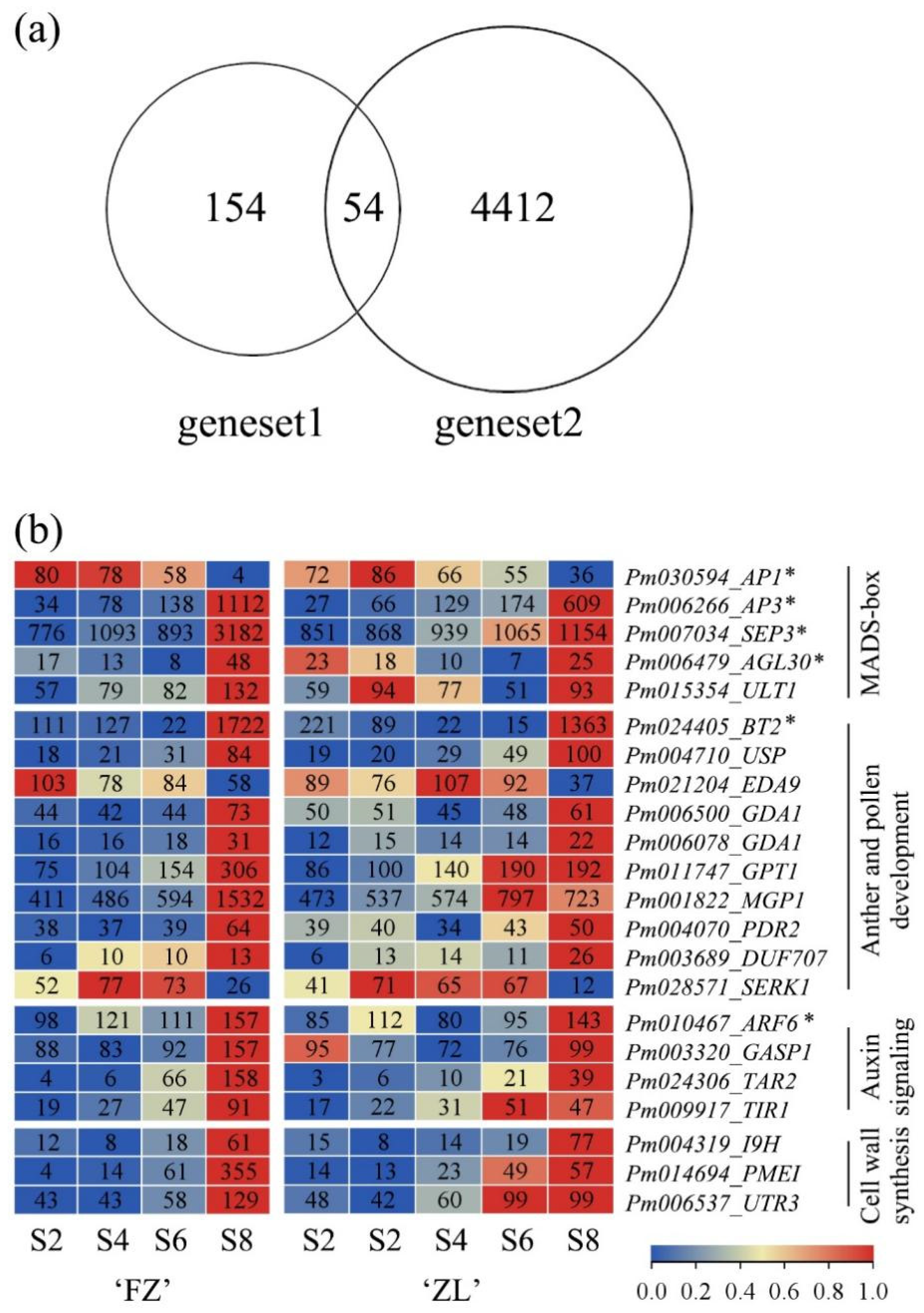
3.3. Dap-Seq Combined with WGCNA to Screen Downstream Key Genes of PmMYB21
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.Y. Origin and history. In Chinese Mei Flowers; Chen, J.Y., Ed.; Hainan Publishing House: Haikou, China, 1996; p. 2. [Google Scholar]
- Yuan, X.; Ma, K.F.; Zhang, M.H.; Zhang, Z.Y.; Zhang, Q.X. How does an early flowering cultivar of Prunus mume arrange its bud development in late winter? Acta Hortic. 2020, 1, 15–20. [Google Scholar] [CrossRef]
- Nishiyama, S.; Matsushita, M.C.; Yamane, H.; Honda, C.; Okada, K.; Tamada, Y.; Moriya, S.; Tao, R. Functional and expressional analyses of apple FLC-like in relation to dormancy progress and flower bud development. Tree Physiol. 2021, 41, 562–570. [Google Scholar] [CrossRef]
- Falavigna, V.D.S.; Guitton, B.; Costes, E.; Andrés, F. I Want to (Bud) Break Free: The Potential Role of DAM and SVP-Like Genes in Regulating Dormancy Cycle in Temperate Fruit Trees. Front. Plant Sci. 2018, 9, 1990. [Google Scholar] [CrossRef]
- Yamane, H.; Kashiwa, Y.; Ooka, T.; Tao, R.; Yonemori, K. Suppression Subtractive Hybridization and Differential Screening Reveals Endodormancy-associated Expression of an SVP/AGL24-type MADS-box Gene in Lateral Vegetative Buds of Japanese Apricot. J. Am. Soc. Hort. Sci. 2008, 133, 708–716. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, M.; Ma, K.F.; Wang, J.; Zhang, Q.X. Function and Expression Regulation Analysis of Prunus mume PmMYB21 During Filament Elongation. Acta Hortic. Sin. 2023, 50, 1048–1062. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, C.; Zuo, J.; Yan, X.; Zhang, J.; Bao, M. Identification and expression analysis of R2R3-MYB transcription factors in Prunus mume under cold treatment. J. Beijing For. Univ. 2015, 37, 5. [Google Scholar]
- Cheng, H.; Song, S.; Xiao, L.; Soo, H.M.; Cheng, Z.; Xie, D.; Peng, J. Gibberellin acts through jasmonate to control the expression of MYB21, MYB24, and MYB57 to promote stamen filament growth in Arabidopsis. PLoS Genet. 2009, 5, e1000440. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Mandaokar, A.; Thines, B.; Shin, B.; Lange, B.M.; Choi, G.; Koo, Y.J.; Yoo, Y.J.; Choi, Y.D.; Choi, G.; Browse, J. Transcriptional regulators of stamen development in Arabidopsis identified by transcriptional profiling. Plant J. 2006, 46, 984–1008. [Google Scholar] [CrossRef]
- Shin, B.; Choi, G.; Yi, H.; Yang, S.; Cho, I.; Kim, J.; Lee, S.; Paek, N.C.; Kim, J.H.; Song, P.S.; et al. AtMYB21, a gene encoding a flower-specific transcription factor, is regulated by COP1. Plant J. 2002, 30, 23–32. [Google Scholar] [CrossRef]
- Song, S.; Qi, T.; Huang, H.; Ren, Q.; Wu, D.; Chang, C.; Peng, W.; Liu, Y.; Peng, J.; Xie, D. The Jasmonate-ZIM domain proteins interact with the R2R3-MYB transcription factors MYB21 and MYB24 to affect Jasmonate-regulated stamen development in Arabidopsis. Plant Cell. 2011, 23, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- López-Vidriero, I.; Godoy, M.; Grau, J.; Peñuelas, M.; Solano, R.; Franco-Zorrilla, J.M. DNA features beyond the transcription factor binding site specify target recognition by plant MYC2-related bHLH proteins. Plant Commun. 2021, 2, 100232. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Lillo, G.; Pérez-Reyes, W.; Riveros, A.; Lillo-Carmona, V.; Rothkegel, K.; Álvarez, J.M.; Blanco-Herrera, F.; Pedreschi, R.; Campos-Vargas, R.; Meneses, C. Transcriptome and Gene Regulatory Network Analyses Reveal New Transcription Factors in Mature Fruit Associated with Harvest Date in Prunus persica. Plants 2022, 11, 3473. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell. 2016, 165, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, A.; O’Malley, R.C.; Huang, S.C.; Galli, M.; Nery, J.R.; Gallavotti, A.; Ecker, J.R. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat. Protoc. 2017, 12, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Vasimuddin, M.; Sanchit, M.; Heng, L.; Srinivas, A. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; Available online: https://ieeexplore.ieee.org/document/8820962 (accessed on 2 September 2019).
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Machanick, P.; Bailey, T.L. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics. 2011, 27, 1696–1697. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Yuan, X.; Ma, K.; Zhang, M.; Wang, J.; Zhang, Q. Integration of Transcriptome and Methylome Analyses Provides Insight Into the Pathway of Floral Scent Biosynthesis in Prunus mume. Front. Genet. 2021, 12, 779557. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Szécsi, J.; Joly, C.; Bordji, K.; Varaud, E.; Cock, J.M.; Dumas, C.; Bendahmane, M. BIGPETALp, a bHLH transcription factor is involved in the control of Arabidopsis petal size. EMBO J. 2006, 25, 3912–3920. [Google Scholar] [CrossRef]
- Mei, F.; Chen, B.; Du, L.; Li, S.; Zhu, D.; Chen, N.; Zhang, Y.; Li, F.; Wang, Z.; Cheng, X.; et al. A gain-of-function allele of a DREB transcription factor gene ameliorates drought tolerance in wheat. Plant Cell 2022, 34, 4472–4494. [Google Scholar] [CrossRef]
- Liu, W.; Zheng, T.; Qiu, L.; Guo, X.; Li, P.; Yong, X.; Li, L.; Ahmad, S.; Wang, J.; Cheng, T.; et al. A 49-bp deletion of PmAP2L results in a double flower phenotype in Prunus mume. Hortic. Res. 2024, 11, uhad278. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Shan, W.; Yang, T.W.; Wu, C.J.; Liu, X.C.; Chen, J.Y.; Lu, W.J.; Li, Z.G.; Deng, W.; Kuang, J.F. MaMYB4 is a negative regulator and a substrate of RING-type E3 ligases MaBRG2/3 in controlling banana fruit ripening. Plant J. 2022, 110, 1651–1669. [Google Scholar] [CrossRef] [PubMed]
- Dubois, A.; Remay, A.; Raymond, O.; Balzergue, S.; Chauvet, A.; Maene, M.; Pécrix, Y.; Yang, S.H.; Jeauffre, J.; Thouroude, T.; et al. Genomic approach to study floral development genes in Rosa sp. PLoS ONE 2011, 6, e28455. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Gupta, K.; Pathania, S.; Swarnkar, M.K.; Rattan, U.K.; Singh, G.; Sharma, R.K.; Singh, A.K. Chilling Affects Phytohormone and Post-Embryonic Development Pathways during Bud Break and Fruit Set in Apple (Malus domestica Borkh.). Sci. Rep. 2017, 7, 42593. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, J.; Bracha-Drori, K.; Yalovsky, S.; Ito, T.; Yu, H. Specification of Arabidopsis floral meristem identity by repression of flowering time genes. Development 2007, 134, 1901–1910. [Google Scholar] [CrossRef]
- Li, C.; Chen, L.; Fan, X.; Qi, W.; Ma, J.; Tian, T.; Zhou, T.; Ma, L.; Chen, F. MawuAP1 promotes flowering and fruit development in the basal angiosperm Magnolia wufengensis (Magnoliaceae). Tree Physiol. 2020, 40, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, Q.; Sun, L.; Du, D.; Cheng, T.; Pan, H.; Yang, W.; Wang, J. Genome-wide identification, characterisation and expression analysis of the MADS-box gene family in Prunus mume. Mol. Genet. Genom. 2014, 289, 903–920. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Krizek, B.A.; Meyerowitz, E.M. Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc. Natl. Acad. Sci. USA 1996, 93, 4793–4798. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, S.; Ditta, G.S.; Baumann, E.; Wisman, E.; Yanofsky, M.F. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef]
- Honma, T.; Goto, K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 2001, 409, 525–529. [Google Scholar] [CrossRef]
- Theissen, G. Development of floral organ identity: Stories from the MADS house. Curr. Opin. Plant Biol. 2001, 4, 75–85. [Google Scholar] [CrossRef]
- Jack, T.; Fox, G.L.; Meyerowitz, E.M. Arabidopsis homeotic gene APETALA3 ectopic expression: Transcriptional and posttranscriptional regulation determine floral organ identity. Cell 1994, 76, 703–716. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.J.; Ahn, J.H. Role of SEPALLATA3 (SEP3) as a downstream gene of miR156-SPL3-FT circuitry in ambient temperature-responsive flowering. Plant Signal Behav. 2012, 7, 1151–1154. [Google Scholar] [CrossRef]
- Honys, D.; Twell, D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 2004, 5, R85. [Google Scholar] [CrossRef]
- Adamczyk, B.J.; Fernandez, D.E. MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 2009, 149, 1713–1723. [Google Scholar] [CrossRef]
- Li, Z.; Luo, D.; Tang, M.; Cao, S.; Pan, J.; Zhang, W.; Hu, Y.; Yue, J.; Huang, Z.; Li, R.; et al. Integrated Methylome and Transcriptome Analysis Provides Insights into the DNA Methylation Underlying the Mechanism of Cytoplasmic Male Sterility in Kenaf (Hibiscus cannabinus L.). Int. J. Mol. Sci. 2022, 23, 6864. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, P.; Ellis, C.M.; Weber, H.; Ploense, S.E.; Barkawi, L.S.; Guilfoyle, T.J.; Hagen, G.; Alonso, J.M.; Cohen, J.D.; Farmer, E.E.; et al. Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development 2005, 132, 4107–4118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.X.; Yin, X.X.; Li, S.; Peng, Y.T.; Yan, X.L.; Chen, C.; Hassan, B.; Zhou, S.X.; Pu, M.; Zhao, J.H.; et al. miR167d-ARFs Module Regulates Flower Opening and Stigma Size in Rice. Rice 2022, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Robert, H.S.; Quint, A.; Brand, D.; Vivian-Smith, A.; Offringa, R. BTB and TAZ domain scaffold proteins perform a crucial function in Arabidopsis development. Plant J. 2009, 58, 109–121. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.; He, R.; Zhang, H.; Liu, D.; Liu, D.; Niu, Z.; Zhang, Y.; Xia, X. Deciphering the Regulatory Mechanism of PmMYB21 in Early Flowering of Prunus mume through Dap-Seq and WGCNA Analysis. Forests 2024, 15, 1300. https://doi.org/10.3390/f15081300
Yuan X, He R, Zhang H, Liu D, Liu D, Niu Z, Zhang Y, Xia X. Deciphering the Regulatory Mechanism of PmMYB21 in Early Flowering of Prunus mume through Dap-Seq and WGCNA Analysis. Forests. 2024; 15(8):1300. https://doi.org/10.3390/f15081300
Chicago/Turabian StyleYuan, Xi, Ran He, Hui Zhang, Dongyan Liu, Donghuan Liu, Zhihong Niu, Yu Zhang, and Xinli Xia. 2024. "Deciphering the Regulatory Mechanism of PmMYB21 in Early Flowering of Prunus mume through Dap-Seq and WGCNA Analysis" Forests 15, no. 8: 1300. https://doi.org/10.3390/f15081300