
Genetic Diversity and Population Structural Analysis Reveal the Unique Genetic Composition of Populus tomentosa Elite Trees


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. Primer Screening for the Polymorphism
2.3. SSR Genotyping
2.4. Data Analysis
3. Results
3.1. Polymorphic SSR Primers
3.2. Microsatellite Polymorphisms
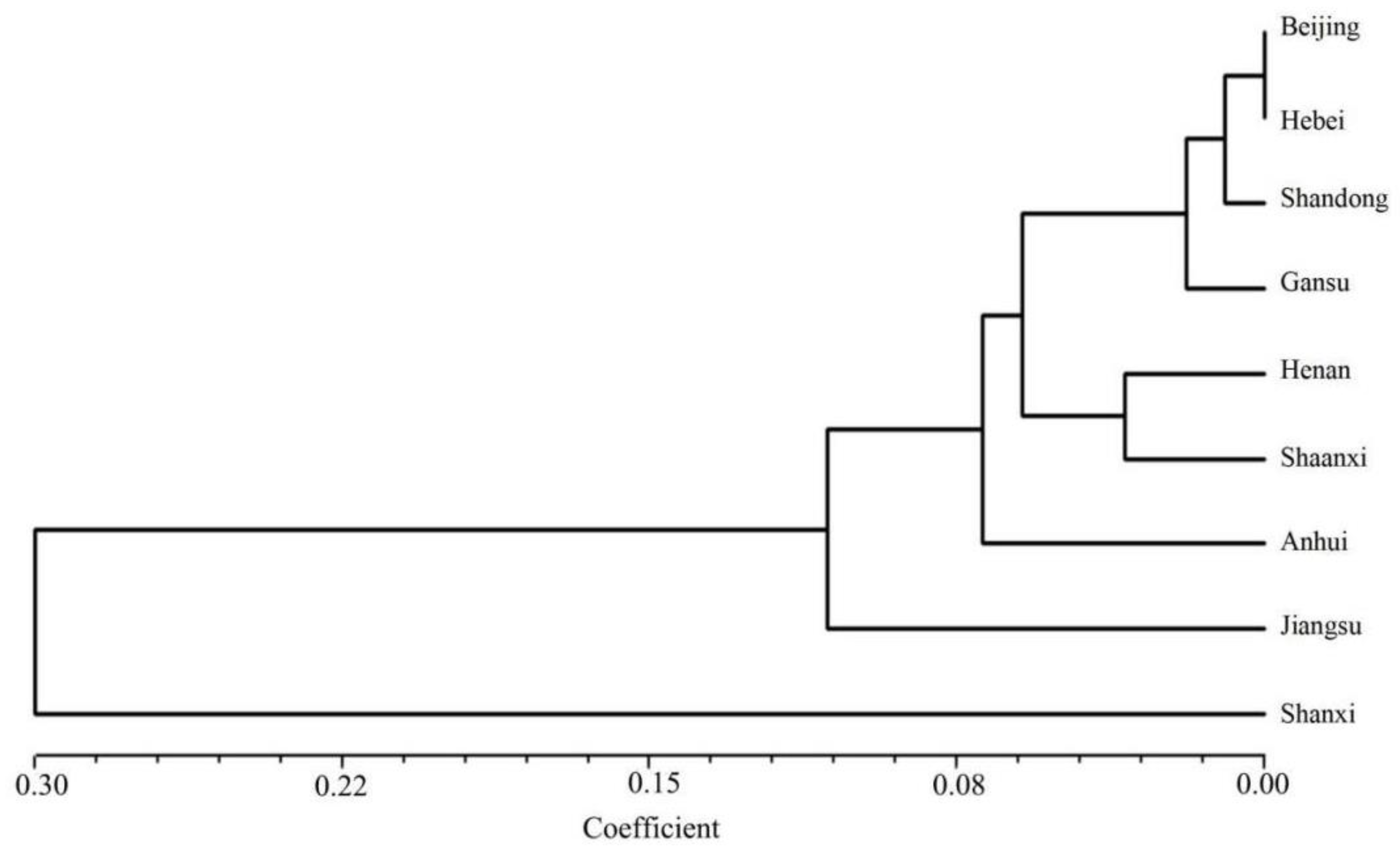
3.3. Population Genetic Diversity and Genetic Differentiation
3.4. Population Genetic Structure
4. Discussion
4.1. Genetic Parameters of the P. tomentosa Elite Trees
4.2. Population Diversity and Structure of the P. tomentosa Elite Trees
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New For. 1992, 6, 95–124. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Effects of Life History Traits on Genetic Diversity in Plant Species. Philosophical Transactions of the Royal Society of London. Ser. B Biol. Sci. 1997, 351, 1291–1298. [Google Scholar] [CrossRef]
- Neel, M.C.; Ellstrand, N.C. Conservation of Genetic Diversity in the Endangered Plant Eriogonum ovalifolium Var. Vineum (Polygonaceae). Conserv. Genet. 2003, 4, 337–352. [Google Scholar] [CrossRef]
- Tsarev, A.P.; Tsareva, R.P.; Tsarev, V.A.; Miligula, E.; Lenchenkova, O. Introduced poplar varieties and new hybrids for protective afforestation. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Voronezh, Russia, 23 October 2020; Volume 595. [Google Scholar] [CrossRef]
- Burgess, P.J.; Incoll, L.D.; Corry, D.T.; Beaton, A.; Hart, B.J. Poplar (Populus Spp.) Growth and Crop Yields in a Silvoarable Experiment at Three Lowland Sites in England. Agrofor. Syst. 2005, 63, 157–169. [Google Scholar] [CrossRef]
- Niemczyk, M.; Kaliszewski, A.; Jewiarz, M.; Wróbel, M.; Mudryk, K. Productivity and Biomass Characteristics of Selected Poplar (Populus spp.) Cultivars under the Climatic Conditions of Northern Poland. Biomass Bioenergy 2018, 111, 46–51. [Google Scholar] [CrossRef]
- Brunner, A.M.; Busov, V.B.; Strauss, S.H. Poplar Genome Sequence: Functional Genomics in an Ecologically Dominant Plant Species. Trends Plant Sci. 2004, 9, 49–56. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G. Populus: Arabidopsis for Forestry. Do We Need a Model Tree? Ann. Bot. 2002, 90, 681–689. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhang, X.; Yao, W.; Gao, Y.; Zhao, K.; Guo, Q.; Zhou, B.; Jiang, T. Genome-Wide Identification and Expression Analysis of the Xyloglucan Endotransglucosylase/Hydrolase Gene Family in Poplar. BMC Genom. 2021, 22, 1–13. [Google Scholar] [CrossRef]
- Li, J.; Gao, K.; Khan, W.U.; Yang, X.; Yang, X.; Zhao, T.; Chen, Z.; An, X. Genome-wide analysis of the poplar NF-Y gene family and its expression in floral bud development of Populus tomentosa. Trees 2019, 34, 285–296. [Google Scholar] [CrossRef]
- Wang, D.; Meng, S.; Su, W.; Bao, Y.; Lu, Y.; Yin, W.; Liu, C.; Xia, X. Genome-Wide Analysis of Multiple Organellar RNA Editing Factor Family in Poplar Reveals Evolution and Roles in Drought Stress. Int. J. Mol. Sci. 2019, 20, 1425. [Google Scholar] [CrossRef]
- Carretero-Paulet, L.; Galstyan, A.; Roig-Villanova, I.; Martínez-García, J.F.; Bilbao-Castro, J.R.; Robertson, D.L. Genome-Wide Classification and Evolutionary Analysis of the bHLH Family of Transcription Factors in Arabidopsis, Poplar, Rice, Moss, and Algae. Plant Physiol. 2010, 153, 1398–1412. [Google Scholar] [CrossRef]
- Han, H.L. Study on the Ecological Function and Optimization Countermeasures of Street Trees in Urban Main Roads. Master’s Thesis, Shandong Jianzhu University, Jinan, China, 2022. [Google Scholar] [CrossRef]
- Kang, X.Y.; Zhu, Z.T. Status and role of triploid white poplar in paper pulp production in China. J. Beijing For. Univ. 2002, 24 (Suppl. S1), 51–56. [Google Scholar]
- Zhu, Z.T. Genetic Improvement of Populus tomentosa Carr.; China Forestry Publishing House: Beijing, China, 2006. [Google Scholar]
- Kang, X.Y. Thinking and practices for strategy on a new round genetic improvement of Populus tomentosa. J. Beijing For. Univ. 2016, 38, 1–8. [Google Scholar] [CrossRef]
- Bai, F.Y.; Zeng, Q.Q.; Kang, N.; Suo, Y.J.; Liao, T.; Zhang, P.D.; Kang, X.Y. Ploidy level and contrast analysis of the traits for superior trees of Populus tomentosa Carr. in gene pool. J. Beijing For. Univ. 2015, 37, 113–119. [Google Scholar] [CrossRef]
- Moose, S.P.; Mumm, R.H. Molecular Plant Breeding as the Foundation for 21st Century Crop Improvement. Plant Physiol. 2008, 147, 969–977. [Google Scholar] [CrossRef]
- Du, Q.; Wang, B.; Wei, Z.; Zhang, D.; Li, B. Genetic Diversity and Population Structure of Chinese White Poplar (Populus tomentosa) Revealed by SSR Markers. J. Hered. 2012, 103, 853–862. [Google Scholar] [CrossRef]
- Quan, M.; Liu, X.; Xiao, L.; Chen, P.; Song, F.; Lu, W.; Song, Y.; Zhang, D. Transcriptome Analysis and Association Mapping Reveal the Genetic Regulatory Network Response to Cadmium Stress in Populus tomentosa. J. Exp. Bot. 2021, 72, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chu, Y.; Ding, C.; Su, X.; Huang, Q. Genetic Diversity and Population Structure of Black Cottonwood (Populus deltoides) Revealed Using Simple Sequence Repeat Markers. BMC Genet 2020, 21, 2. [Google Scholar] [CrossRef]
- Drost, D.R.; Novaes, E.; Boaventura-Novaes, C.; Benedict, C.I.; Brown, R.S.; Yin, T.; Tuskan, G.A.; Kirst, M. A Microarray-Based Genotyping and Genetic Mapping Approach for Highly Heterozygous Outcrossing Species Enables Localization of a Large Fraction of the Unassembled Populus trichocarpa Genome Sequence. Plant J. 2009, 58, 1054–1067. [Google Scholar] [CrossRef]
- Wei, Z.; Du, Q.; Zhang, J.; Li, B.; Zhang, D. Genetic Diversity and Population Structure in Chinese Indigenous Poplar (Populus Simonii) Populations Using Microsatellite Markers. Plant Mol. Biol. Rep. 2012, 31, 620–632. [Google Scholar] [CrossRef]
- Decroocq, V.; Favé, M.G.; Hagen, L.; Bordenave, L.; Decroocq, S. Development and Transferability of Apricot and Grape EST Microsatellite Markers across Taxa. Theor. Appl. Genet. 2003, 106, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, D.R.; Kumar, R.; Maurya, A.; Semwal, D.P.; Rathi, R.S.; Gautam, R.K.; Trivedi, A.K.; Bishnoi, S.K.; Ahlawat, S.P.; Singh, K.; et al. SSR and SNP Marker-Based Investigation of Indian Rice Landraces in Relation to Their Genetic Diversity, Population Structure, and Geographical Isolation. Agriculture 2023, 13, 823. [Google Scholar] [CrossRef]
- Han, Z.; Han, Q.; Xia, Y.; Geng, X.; Du, K.; Yang, J.; Kang, X. Construction of a Breeding Parent Population of Populus Tomentosa Based on SSR Genetic Distance Analysis. Sci. Rep. 2020, 10, 18573. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.S.; Wen, G.M.; Gou, S.Y.; Li, B.P. Selective breeding of clone resistant to cold on Populus tomentosa. Shanxi For. Sci. Technol. 1998, 12, 4. [Google Scholar]
- Han, Z.Q. Study on the Selection Strategy of Populus tomentosa Breeding Parents Based on SSR Markers; Beijing Forestry University: Beijing, China, 2018. [Google Scholar] [CrossRef]
- Yeh, F.C.; Yang, R.; Boyle, T. POPGENE Version 1.32 Microsoft Windows-Based Freeware for Populations Genetic Analysis; University of Alberta: Edmonton, AB, Canada, 1999. [Google Scholar]
- Schuelke, M. An Economic Method for the Fluorescent Labeling of PCR Fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic Analysis in Excel. Population Genetic Software for Teaching and Research—An Update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Jost, L. Entropy and Diversity. Oikos 2006, 113, 363–375. [Google Scholar] [CrossRef]
- Sievert, C. Interactive Web-Based Data Visualization with R, Plotly, and Shiny; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: Columbia, NY, USA, 1987. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software STRUCTURE: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. Structure Harvester: A Website and Program for Visualizing STRUCTURE Output and Implementing the Evanno Method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Ellegren, H.; Galtier, N. Determinants of Genetic Diversity. Nat. Rev. Genet. 2016, 17, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Falk, D.A.; Holsinger, K.E. Genetics and Conservation of Rare Plants; Oxford University Press: Oxford, UK, 1991. [Google Scholar]
- Yao, J.X.; Mao, X.H.; Li, S.W.; Liu, X.L.; Wu, D. Genetic diversity of germplasm resources of Leuce based on SSR fluorescent marker. J. Beijing For. Univ. 2018, 40, 92–100. [Google Scholar] [CrossRef]
- Fang, J.Y.; Shen, Z.H.; Tang, Z.Y.; Wang, Z.H. The protocol for the survey plan for plant species diversity of China’s mountains. Biodivers. Sci. 2004, 12, 5–9. [Google Scholar] [CrossRef]
- Lee, K.M.; Kim, Y.Y.; Hyun, J.O. Genetic Variation in Populations of Populus Davidiana Dode Based on Microsatellite Marker Analysis. Genes Genom. 2011, 33, 163–171. [Google Scholar] [CrossRef]
- Makrem, A.; Najeh, B.F.; Laarbi, K.M.; Mohamed, B. Genetic Diversity in Tunisian Ceratonia siliqua L. (Caesalpinioideae) Natural Populations. Genet Resour. Crops Evol. 2006, 53, 1501–1511. [Google Scholar] [CrossRef]
- Chapuis, M.-P.; Estoup, A. Microsatellite Null Alleles and Estimation of Population Differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef]
- Solis, W.; Fuchs, E. Effective Gene Flow Patterns across a Fragmented Landscape in Southern Costa Rica for Symphonia globulifera (Clusiaceae); a Species with Mobile Seed and Pollen Dispersers. Rev. Biol. Trop. 2019, 67, 95–111. [Google Scholar] [CrossRef]
- Martins, K.; Kimura, R.K.; Francisconi, A.F.; Gezan, S.; Kainer, K.; Christianini, A.V. The Role of Very Small Fragments in Conserving Genetic Diversity of a Common Tree in a Hyper Fragmented Brazilian Atlantic Forest Landscape. Conserv. Genet. 2016, 17, 509–520. [Google Scholar] [CrossRef]
- Aguiar, B.I.; Freitas, M.L.M.; Zannato, A.S.; Tambarussi, E.V.; Moraes, M.L.T.; Ambrosano, M.N.; Pereira, L.C.S.M.; Gandara, F.B.; Kageyama, P.Y.; Sebbenn, A.M. The Effects of Pollen Dispersal and Mating Pattern on Inbreeding Depression and Hybrid Vigor in Balfourodendron riedelianum (Engl.) Engl. (Rutaceae). Conserv. Genet. 2020, 21, 305–317. [Google Scholar] [CrossRef]
- Griffin, A.R.; Potts, B.M.; Vaillancourt, R.E.; Bell, J.C. Life Cycle Expression of Inbreeding Depression in Eucalyptus Regnans and Inter-Generational Stability of Its Mixed Mating System. Ann. Bot. 2019, 124, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Eckert, C.G.; Samis, K.E.; Lougheed, S.C. Genetic Variation across Species’ Geographical Ranges: The Central-Marginal Hypothesis and Beyond. Mol. Ecol. 2008, 17, 1170–1188. [Google Scholar] [CrossRef] [PubMed]
- Lv, W. Populus simonii in North of China. For. Sci. Technol. 2002, 6, 31–32. [Google Scholar] [CrossRef]
- Wei, Z.Z.; Pan, W.; Zhao, X.; Zhang, J.F.; Li, B.; Zhang, D. Morphological and Physiological Genetic Diversity of Populus Simonii in Northeastern and North China. J. Beijing For. Univ. 2010, 32, 8–14. [Google Scholar] [CrossRef]
- Budke, J.C.; Jarenkow, J.A.; de Oliveira-Filho, A.T. Intermediary Disturbance Increases Tree Diversity in Riverine Forest of Southern Brazil. Biodivers Conserv. 2010, 19, 2371–2387. [Google Scholar] [CrossRef]
- Wu, J.G.; Lv, J.J.; Ai, L. The impacts of climate change on the biodiversity: Vulnerability and Adaptation. Ecol. Environ. Sci. 2009, 18, 693–703. [Google Scholar] [CrossRef]
- Liu, X.; Ma, Y.; Wan, Y.; Li, Z.; Ma, H. Genetic Diversity of Phyllanthus Emblica From Two Different Climate Type Areas. Front. Plant Sci. 2020, 11, 580812. [Google Scholar] [CrossRef] [PubMed]
- Posselt, D.J. Markov Chain Monte Carlo Methods: Theory and Applications. In Data Assimilation for Atmospheric, Oceanic and Hydrologic Applications (Vol. II); Park, S.K., Xu, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 59–87. [Google Scholar] [CrossRef]
- Jacob, P.E.; O’Leary, J.; Atchadé, Y.F. Unbiased Markov Chain Monte Carlo with Couplings. arXiv 2019, arXiv:1708.03625. [Google Scholar] [CrossRef]
- Swarup, S.; Cargill, E.J.; Crosby, K.; Flagel, L.; Kniskern, J.; Glenn, K.C. Genetic Diversity Is Indispensable for Plant Breeding to Improve Crops. Crops Sci. 2021, 61, 839–852. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | SSR Primer Sequence (5′-3′) | Chromosome | Motif | Expected Size (bp) |
|---|---|---|---|---|
| LG_III-2 | Forward: ATTGATTATATTTGCCGCAT Reverse: TGGACATCTCACTACCTTCC | Chr03 | AT/TA | 180, 186 |
| GCPM_2570-1 | Forward: AACCCACTTCCTCTCTCTGT Reverse: TGAGACTTCCGACTCGTAG | Chr01 | CT | 229, 233, 237 |
| ORPM_197 | Forward: GTCAGTTTGCCCTCTTCGTC Reverse: TGAGGGCGTCTCCTCTTTTA | Chr06 | GA | 206, 208 |
| PMGC_2140 | Forward: GCTGTCAGAATCAAACACTTC Reverse: AAGCAGATAACTAAGACATGCC | Chr07 | GA | 174, 176 |
| PMGC_223 | Forward: CGATGAGGTTGAAGAAGTCG Reverse: ATATATGTACCGGCACGCCAC | Chr02 | CTT | 185, 191 |
| LG_VIII-3 | Forward: ATCCGACTTCGATATCTTCA Reverse: CTACCTGAAACACAGGAAGC | Chr08 | AG/TC | 238, 254 |
| GCPM_112-1 | Forward: TTAGAGGAGAGAACTGCTGC Reverse: TGGTCTGCAACACAAGATT | Chr16 | GT | 127, 131, 133 |
| GCPM_1411-1 | Forward: TCAACGACTTTTTCATTGTG Reverse: AGCATTCTTGCTGGTGTTAT | Chr02 | TGC | 232, 235, 238 |
| GCPM_1153-1 | Forward: TTCCTTTCACACAATGACAA Reverse: TTTAAAAACTGGGTCCGTAA | Chr11 | CTT | 165, 171, 174, 180 |
| LG_XVI-6 | Forward: ATAGCGATCATCAAAGGAAA Reverse: AAATATTCATGTGGAGGCAC | Chr16 | AG/TC | 116, 130 |
| PMGC_2818 | Forward: AAGCTTCATCGTCCTGCTTG Reverse: CGTATCAATTCACGACTCTCG | Chr02 | GA | 138, 140 |
| LG_XVI-7 | Forward: ACAAATCAAAGTCACAGCCT Reverse: ATAGTGTTCAATCGGACCTG | Chr16 | AG/TC | 352, 362, 364 |
| GCPM_1832-1 | Forward: TTACTTGCTAGCTGCCAATC Reverse: CCTAAAAGTTTGTCTATGCGA | Chr02 | TA | 158, 160 |
| Locus | Na | Ne | I | PIC | Ho | He | Fis | Fit | Fst | Nm |
|---|---|---|---|---|---|---|---|---|---|---|
| LG_III-2 | 7 | 2.461 | 1.292 | 0.569 | 0.374 | 0.594 | 0.039 | 0.248 | 0.217 | 0.900 |
| GCPM_2570-1 | 12 | 4.258 | 1.648 | 0.729 | 0.818 | 0.766 | −0.249 | −0.151 | 0.078 | 2.944 |
| ORPM_197 | 3 | 2.068 | 0.874 | 0.453 | 0.111 | 0.517 | 0.643 | 0.683 | 0.113 | 1.970 |
| PMGC_2140 | 10 | 3.005 | 1.498 | 0.636 | 0.319 | 0.668 | 0.349 | 0.460 | 0.171 | 1.215 |
| PMGC_223 | 11 | 4.316 | 1.647 | 0.732 | 0.671 | 0.769 | −0.056 | 0.045 | 0.096 | 2.369 |
| LG_VIII-3 | 9 | 2.672 | 1.314 | 0.594 | 0.024 | 0.626 | 0.938 | 0.951 | 0.206 | 0.965 |
| GCPM_112-1 | 14 | 4.748 | 1.827 | 0.761 | 0.857 | 0.790 | −0.282 | −0.197 | 0.066 | 3.524 |
| GCPM_1411-1 | 15 | 2.896 | 1.451 | 0.623 | 0.379 | 0.655 | 0.576 | 0.619 | 0.102 | 2.199 |
| GCPM_1153-1 | 13 | 2.162 | 1.284 | 0.520 | 0.493 | 0.538 | 0.001 | 0.116 | 0.115 | 1.922 |
| LG_XVI-6 | 4 | 2.369 | 0.996 | 0.503 | 0.867 | 0.578 | −0.658 | −0.613 | 0.027 | 9.024 |
| PMGC_2818 | 12 | 2.460 | 1.115 | 0.511 | 0.835 | 0.594 | −0.331 | −0.260 | 0.054 | 4.398 |
| LG_XVI-7 | 6 | 2.127 | 0.844 | 0.428 | 0.822 | 0.530 | −0.711 | −0.658 | 0.031 | 7.850 |
| GCPM_1832-1 | 9 | 1.375 | 0.675 | 0.266 | 0.094 | 0.273 | 0.704 | 0.729 | 0.083 | 2.763 |
| Mean | 9.6 | 2.840 | 1.266 | 0.563 | 0.513 | 0.608 | 0.020 | 0.123 | 0.105 | 2.125 |
| Pop | N | Na | Ne | I | Ho | He | uHe |
|---|---|---|---|---|---|---|---|
| Beijing (BJ) | 20 | 2.692 | 1.699 | 0.562 | 0.474 | 0.329 | 0.338 |
| Hebei (HB) | 122 | 5.231 | 1.754 | 0.670 | 0.485 | 0.355 | 0.356 |
| Shandong (SD) | 20 | 4.000 | 1.883 | 0.775 | 0.505 | 0.423 | 0.434 |
| Henan (HN) | 113 | 6.000 | 2.440 | 1.068 | 0.492 | 0.547 | 0.550 |
| Shanxi (SX) | 270 | 7.692 | 3.118 | 1.254 | 0.532 | 0.640 | 0.641 |
| Shaanxi (SAX) | 54 | 5.538 | 2.736 | 1.128 | 0.557 | 0.582 | 0.588 |
| Mean | 98.7 | 5.192 | 2.272 | 0.910 | 0.508 | 0.479 | 0.485 |
| Source | df | SS | MS | Variance Estimated | % | F-Statistic | p-Values |
|---|---|---|---|---|---|---|---|
| Among population | 8 | 552.786 | 69.098 | 0.572 | 14 | Fst = 0.136 | 0.001 |
| Among individual | 614 | 2418.324 | 3.939 | 0.316 | 8 | Fis = 0.087 | 0.001 |
| Within individual | 623 | 2060.000 | 3.307 | 3.307 | 79 | Fit = 0.212 | 0.001 |
| Total | 1245 | 5031.111 | - | 4.195 | 100 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, B.; Ma, L.; Du, J.; Zhang, P. Genetic Diversity and Population Structural Analysis Reveal the Unique Genetic Composition of Populus tomentosa Elite Trees. Forests 2024, 15, 1377. https://doi.org/10.3390/f15081377
Kong B, Ma L, Du J, Zhang P. Genetic Diversity and Population Structural Analysis Reveal the Unique Genetic Composition of Populus tomentosa Elite Trees. Forests. 2024; 15(8):1377. https://doi.org/10.3390/f15081377
Chicago/Turabian StyleKong, Bo, Lexun Ma, Jiahua Du, and Pingdong Zhang. 2024. "Genetic Diversity and Population Structural Analysis Reveal the Unique Genetic Composition of Populus tomentosa Elite Trees" Forests 15, no. 8: 1377. https://doi.org/10.3390/f15081377