
Construction of the First High-Density Genetic Linkage Map and QTL Mapping of Shikimic Acid Content in Liquidambar


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Shikimic Acid Content Detection
2.2. DNA Extraction, Library Construction and Sequencing
2.3. Data Analysis and Linkage Map Construction
2.4. QTL Mapping
3. Results
3.1. Preliminary Identification of Hybrid Populations
3.2. Analysis of WGR Data
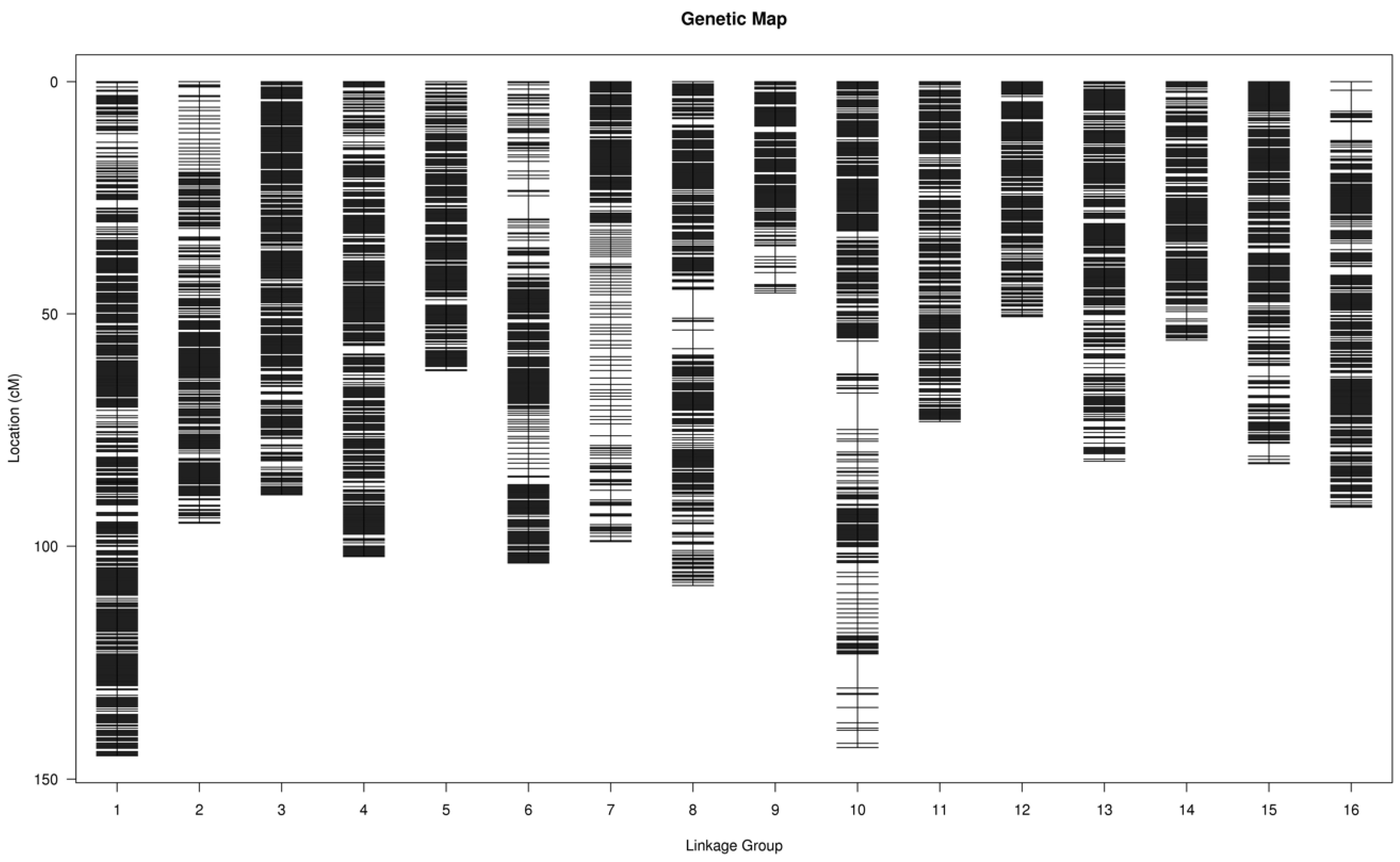
3.3. Construction of Genetic Linkage Map
3.4. QTL Mapping for Shikimic Acid Content
3.5. Potential Candidate Gene Mining
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| QTL | Quantitative trait locus |
| cM | Centimorgan |
| LG | Linkage group |
| SNP | Single nucleotide polymorphism |
| MAS | Molecular marker-assisted selection |
| RAPD | Random amplified polymorphic DNA |
| RFLP | Restriction fragment length polymorphism |
| AFLP | Amplified fragment length polymorphism |
| SSR | Simple sequence repeats |
| SLAF | Specific-locus amplified fragment |
| RRGS | Reduced Representation Genome Sequencing |
| WGR | Whole-Genome Resequencing |
| CTAB | Cetyltrimethylammonium bromide |
| HPLC | High-performance liquid chromatography |
| LOD | Logarithm of odds |
| GO | Gene ontology |
| MEP | Methylerythritol phosphate |
| DXS | 1-deoxyxylulose-5-phosphate synthase |
References
- Brand, M.H.; Lineberger, R.D. Micropropagation of American Sweetgum (Liquidambar styraciflua L.). In High-Tech and Micropropagation II. Biotechnology in Agriculture and Forestry; Bajaj, Y.P.S., Ed.; Springer: Berlin/Heidelberg, Germany, 1992; Volume 18, pp. 3–24. [Google Scholar]
- Ickert-Bond, S.; Pigg, K.; Wen, J. Comparative infructescence morphology in Liquidambar (Altingiaceae) and its evolutionary significance. Am. J. Bot. 2005, 92, 1234–1255. [Google Scholar] [CrossRef] [PubMed]
- Sutter, E.G. Sweetgum (Liquidambar styraciflua L.). In Trees II. Biotechnology in Agriculture and Forestry; Bajaj, Y.P.S., Ed.; Springer: Berlin/Heidelberg, Germany, 1989; Volume 5, pp. 287–299. [Google Scholar]
- Ďurkovič, J.; Lux, A. Micropropagation with a novel pattern of adventitious rooting in American sweetgum (Liquidambar styraciflua L.). Trees 2010, 24, 491–497. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Y.; Chen, H.; Chen, C.; Liu, Z.; Han, C.; Wu, Q.; Yu, F. Transcriptomic Analyses Reveal Key Genes Involved in Pigment Biosynthesis Related to Leaf Color Change of Liquidambar formosana Hance. Molecules 2022, 27, 5433. [Google Scholar] [CrossRef]
- Sun, R.; Lin, F.; Huang, P.; Zheng, Y. Moderate Genetic Diversity and Genetic Differentiation in the Relict Tree Liquidambar formosana Hance Revealed by Genic Simple Sequence Repeat Markers. Front. Plant Sci. 2016, 7, 1411. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Guan, Y.-J.; Chen, Q.-Z.; Yuan, L.-H.; Xu, Q.-Q.; Zhou, M.-L.; Liu, H.; Lin, W.; Zhang, Z.-D.; Zhou, Z.-L.; et al. Pentacyclic Triterpenes from the resin of Liquidambar formosana have anti-angiogenic properties. Phytochemistry 2021, 184, 112676. [Google Scholar] [CrossRef]
- DeCarlo, A.; Zeng, T.; Dosoky, N.S.; Satyal, P.; Setzer, W.N. The Essential Oil Composition and Antimicrobial Activity of Liquidambar formosana Oleoresin. Plants 2020, 9, 822. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Pan, Y.; Wang, H.; Zhang, Y.; Lei, Q.; Zhu, Z.; Li, H.; Liang, M. Antioxidant activities of Liquidambar formosana Hance leaf extracts. Med. Chem. Res. 2010, 19, 166–176. [Google Scholar] [CrossRef]
- Lingbeck, J.M.; O’Bryan, C.A.; Martin, E.M.; Adams, J.P.; Crandall, P.G. Sweetgum: An ancient source of beneficial compounds with modern benefits. Pharmacogn Rev. 2015, 9, 1–11. [Google Scholar] [CrossRef]
- El-Readi, M.Z.; Eid, H.H.; Ashour, M.L.; Eid, S.Y.; Labib, R.M.; Sporer, F.; Wink, M. Variations of the chemical composition and bioactivity of essential oils from leaves and stems of Liquidambar styraciflua (Altingiaceae). J. Pharm. Pharmacol. 2013, 65, 1653–1663. [Google Scholar] [CrossRef]
- Bochkov, D.V.; Sysolyatin, S.V.; Kalashnikov, A.I.; Surmacheva, I.A. Shikimic acid: Review of its analytical, isolation, and purification techniques from plant and microbial sources. J. Chem. Biol. 2012, 5, 5–17. [Google Scholar] [CrossRef]
- Herrmann, K.M.; Weaver, L.M. THE SHIKIMATE PATHWAY. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 473–503. [Google Scholar] [CrossRef] [PubMed]
- Enrich, L.B.; Scheuermann, M.L.; Mohadjer, A.; Matthias, K.R.; Eller, C.F.; Newman, M.S.; Fujinaka, M.; Poon, T. Liquidambar styraciflua: A renewable source of shikimic acid. Tetrahedron Lett. 2008, 49, 2503–2505. [Google Scholar] [CrossRef]
- Martin, E.; Duke, J.; Pelkki, M.; Clausen, E.C.; Carrier, D.J. Sweetgum (Liquidambar styraciflua L.): Extraction of shikimic acid coupled to dilute acid pretreatment. Appl. Biochem. Biotechnol. 2010, 162, 1660–1668. [Google Scholar] [CrossRef]
- Collard, B.C.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, L.; Lu, W.; Zhong, J.; Du, H.; Liu, P.; Du, Q.; Du, L.; Qing, J. Construction of SNP-Based High-Density Genetic Map Using Genotyping by Sequencing (GBS) and QTL Analysis of Growth Traits in Eucommia ulmoides Oliver. Forests 2022, 13, 1479. [Google Scholar] [CrossRef]
- Nelson, C.D.; Nance, W.L.; Doudrick, R.L. A partial genetic linkage map of slash pine (Pinus elliottii Engelm. var. elliottii) based on random amplified polymorphic DNAs. Theor. Appl. Genet. 1993, 87, 145–151. [Google Scholar] [CrossRef]
- Remington, D.L.; Whetten, R.W.; Liu, B.H.; O’Malley, D.M. Construction of an AFLP genetic map with nearly complete genome coverage in Pinus taeda. Theor. Appl. Genet. 1999, 98, 1279–1292. [Google Scholar] [CrossRef]
- Hirao, T.; Matsunaga, K.; Hirakawa, H.; Shirasawa, K.; Isoda, K.; Mishima, K.; Tamura, M.; Watanabe, A. Construction of genetic linkage map and identification of a novel major locus for resistance to pine wood nematode in Japanese black pine (Pinus thunbergii). BMC Plant Biol. 2019, 19, 424. [Google Scholar] [CrossRef] [PubMed]
- Thamarus, K.A.; Groom, K.; Murrell, J.; Byrne, M.; Moran, G.F. A genetic linkage map for Eucalyptus globulus with candidate loci for wood, fibre, and floral traits. Theor. Appl. Genet. 2002, 104, 379–387. [Google Scholar] [CrossRef]
- Sumathi, M.; Bachpai, V.K.W.; Deeparaj, B.; Mayavel, A.; Dasgupta, M.G.; Nagarajan, B.; Rajasugunasekar, D.; Sivakumar, V.; Yasodha, R. Quantitative trait loci mapping for stomatal traits in interspecific hybrids of Eucalyptus. J. Genet. 2018, 97, 323–329. [Google Scholar] [CrossRef]
- Cervera, M.T.; Storme, V.; Ivens, B.; Gusmão, J.; Liu, B.H.; Hostyn, V.; Van Slycken, J.; Van Montagu, M.; Boerjan, W. Dense genetic linkage maps of three Populus species (Populus deltoides, P. nigra and P. trichocarpa) based on AFLP and microsatellite markers. Genetics 2001, 158, 787–809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, Z.; Yang, K.; Li, B. Genetic mapping in (Populus tomentosa x Populus bolleana) and P. tomentosa Carr. using AFLP markers. Theor. Appl. Genet. 2004, 108, 657–662. [Google Scholar] [CrossRef]
- Lander, E.S. The new genomics: Global views of biology. Science 1996, 274, 536–539. [Google Scholar] [CrossRef]
- Dong, M.; He, Q.; Zhao, J.; Zhang, Y.; Yuan, D.; Zhang, A.J. Genetic Mapping of Prince Rupprecht’s Larch (Larix principis-rupprechtii Mayr) by Specific-Locus Amplified Fragment Sequencing. Genes 2019, 10, 583. [Google Scholar] [CrossRef]
- Chen, X.; Xiong, C.; Lou, Y.; Xu, H.; Cheng, Q.; Sun, S.; Xiao, F. High-Density Genetic Map and QTL Analysis in Cunninghamia lanceolate: Insights into Growth and Wood-Color Traits. Forests 2023, 14, 1591. [Google Scholar] [CrossRef]
- Sun, P.; Jia, H.; Cheng, X.; Zhang, Y.; Li, J.; Zhang, L.; Lu, M.; Zhang, J.; Hu, J. Genetic architecture of leaf morphological and physiological traits in a Populus deltoides ‘Danhong’ × P. simonii ‘Tongliao1’ pedigree revealed by quantitative trait locus analysis. Tree Genet. Genomes 2020, 16, 45. [Google Scholar] [CrossRef]
- Tong, C.; Li, H.; Wang, Y.; Li, X.; Ou, J.; Wang, D.; Xu, H.; Ma, C.; Lang, X.; Liu, G.; et al. Construction of High-Density Linkage Maps of Populus deltoides × P. simonii Using Restriction-Site Associated DNA Sequencing. PLoS ONE 2016, 11, e0150692. [Google Scholar] [CrossRef]
- Peng, Z.; Zhao, C.; Li, S.; Guo, Y.; Xu, H.; Hu, G.; Liu, Z.; Chen, X.; Chen, J.; Lin, S.; et al. Integration of genomics, transcriptomics and metabolomics identifies candidate loci underlying fruit weight in loquat. Hortic. Res. 2022, 9, uhac037. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Z.; Tang, H.; Zhang, Q.; Zhou, G.; Li, X. High-Density Genetic Map Construction and QTL Mapping of Leaf and Needling Traits in Ziziphus jujuba Mill. Front. Plant Sci. 2019, 10, 1424. [Google Scholar] [CrossRef]
- An, Y.; Chen, L.; Tao, L.; Liu, S.; Wei, C. QTL Mapping for Leaf Area of Tea Plants (Camellia sinensis) Based on a High-Quality Genetic Map Constructed by Whole Genome Resequencing. Front. Plant Sci. 2021, 12, 705285. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, Y.; Lai, J.; Wu, J.; Wu, C.; Hu, W.; Wu, X.; Gong, B. The Construction of a High-Density Genetic Map for the Interspecific Cross of Castanea mollissima × C. henryi and the Identification of QTLs for Leaf Traits. Forests 2023, 14, 1684. [Google Scholar] [CrossRef]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yu, Z.; Qi, X.; Wang, Z.; Zheng, Y.; Ren, H.; Liang, S.; Zheng, X. Construction of a High-Density Genetic Map and Identification of Leaf Trait-Related QTLs in Chinese Bayberry (Myrica rubra). Front. Plant Sci. 2021, 12, 675855. [Google Scholar] [CrossRef]
- Chen, S.; Dong, M.; Zhang, Y.; Qi, S.; Liu, X.; Zhang, J.; Zhao, J. Development and Characterization of Simple Sequence Repeat Markers for, and Genetic Diversity Analysis of Liquidambar formosana. Forests 2020, 11, 203. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, G.; Chen, K.; Chen, X.; Hong, Q.; Kan, J. Assessment of fresh star anise (Illicium verum Hook.f.) drying methods for influencing drying characteristics, color, flavor, volatile oil and shikimic acid. Food Chem. 2021, 342, 128359. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ma, C.; Hong, W.; Huang, L.; Liu, M.; Liu, H.; Zeng, H.; Deng, D.; Xin, H.; Song, J.; et al. Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS ONE 2014, 9, e98855. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, J.; Van Ooijen, J.W.; Ooijen, J.; Hoorn, J.; Duin, J.; Jw, V.T.V. MapQTL®6. Software for the Mapping of Quantitative Trait Loci in Experimental Populations of Diploid Species; Kyazma BV: Wageningen, The Netherlands, 2009. [Google Scholar]
- Herrmann, K.M. The Shikimate Pathway: Early Steps in the Biosynthesis of Aromatic Compounds. Plant Cell 1995, 7, 907–919. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, W.; Zhang, J.; Wang, N.; Zhao, Y.; Wang, Y.; Bai, S. Construction of the first high-density genetic linkage map and identification of seed yield-related QTLs and candidate genes in Elymus sibiricus, an important forage grass in Qinghai-Tibet Plateau. BMC Genom. 2019, 20, 861. [Google Scholar] [CrossRef]
- Ferreira, A.; Ferreira, M.; Silva, L.; Cruz, C. Estimating the effects of population size and type on the accuracy of genetic maps. Genet. Mol. Biol. 2006, 29, 187–192. [Google Scholar] [CrossRef]
- Li, H.; Hearne, S.; Bänziger, M.; Li, Z.; Wang, J. Statistical properties of QTL linkage mapping in biparental genetic populations. Heredity 2010, 105, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Deng, G.; Mou, H.; Xu, Y.; Chen, L.; Yang, J.; Zhang, M. A re-sequencing-based ultra-dense genetic map reveals a gummy stem blight resistance-associated gene in Cucumis melo. DNA Res. 2017, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, L.; Wang, J.; Sun, J.; Xia, X.; Geng, X.; Wang, X.; Xu, Z.; Xu, Q. Genome sequencing of rice subspecies and genetic analysis of recombinant lines reveals regional yield- and quality-associated loci. BMC Biol. 2018, 16, 102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Miao, H.; Li, C.; Wei, L.; Duan, Y.; Ma, Q.; Kong, J.; Xu, F.; Chang, S. Ultra-dense SNP genetic map construction and identification of SiDt gene controlling the determinate growth habit in Sesamum indicum L. Sci. Rep. 2016, 6, 31556. [Google Scholar] [CrossRef]
- Zhao, J.; Li, H.; Xu, Y.; Yin, Y.; Huang, T.; Zhang, B.; Wang, Y.; Li, Y.; Cao, Y.; An, W. A consensus and saturated genetic map provides insight into genome anchoring, synteny of Solanaceae and leaf- and fruit-related QTLs in wolfberry (Lycium Linn.). BMC Plant Biol. 2021, 21, 350. [Google Scholar] [CrossRef]
- Yan, F.; Luo, Y.; Bao, J.; Pan, Y.; Wang, J.; Wu, C.; Liu, M. Construction of a highly saturated genetic map and identification of quantitative trait loci for leaf traits in jujube. Front. Plant Sci. 2022, 13, 1001850. [Google Scholar] [CrossRef]
- Liu, D.; Ye, Y.; Tang, R.; Gong, Y.; Chen, S.; Zhang, C.; Mei, P.; Chen, J.; Chen, L.; Ma, C. High-density genetic map construction and QTL mapping of a zigzag-shaped stem trait in tea plant (Camellia sinensis). BMC Plant Biol. 2024, 24, 382. [Google Scholar] [CrossRef]
- Jiang, Y.; Luo, H.; Yu, B.; Ding, Y.; Kang, Y.; Huang, L.; Zhou, X.; Liu, N.; Chen, W.; Guo, J.; et al. High-Density Genetic Linkage Map Construction Using Whole-Genome Resequencing for Mapping QTLs of Resistance to Aspergillus flavus Infection in Peanut. Front. Plant Sci. 2021, 12, 745408. [Google Scholar] [CrossRef]
- Jiang, J.; Fan, X.; Zhang, Y.; Tang, X.; Li, X.; Liu, C.; Zhang, Z. Construction of a High-Density Genetic Map and Mapping of Firmness in Grapes (Vitis vinifera L.) Based on Whole-Genome Resequencing. Int. J. Mol. Sci. 2020, 21, 797. [Google Scholar] [CrossRef]
- Guan, W.; Ke, C.; Tang, W.; Jiang, J.; Xia, J.; Xie, X.; Yang, M.; Duan, C.; Wu, W.; Zheng, Y. Construction of a High-Density Recombination Bin-Based Genetic Map Facilitates High-Resolution Mapping of a Major QTL Underlying Anthocyanin Pigmentation in Eggplant. Int. J. Mol. Sci. 2022, 23, 10258. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Watanabe, M.; Hoefgen, R.; Fernie, A.R. Shikimate and phenylalanine biosynthesis in the green lineage. Front. Plant Sci. 2013, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Adams, Z.P.; Ehlting, J.; Edwards, R. The regulatory role of shikimate in plant phenylalanine metabolism. J. Theor. Biol. 2019, 462, 158–170. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. Auxin biosynthesis: A simple two-step pathway converts tryptophan to indole-3-acetic acid in plants. Mol. Plant 2012, 5, 334–338. [Google Scholar] [CrossRef]
- Cao, X.; Yang, H.; Shang, C.; Ma, S.; Liu, L.; Cheng, J. The Roles of Auxin Biosynthesis YUCCA Gene Family in Plants. Int. J. Mol. Sci. 2019, 20, 6343. [Google Scholar] [CrossRef]
- Di, D.W.; Wu, L.; Zhang, L.; An, C.W.; Zhang, T.Z.; Luo, P.; Gao, H.H.; Kriechbaumer, V.; Guo, G.Q. Functional roles of Arabidopsis CKRC2/YUCCA8 gene and the involvement of PIF4 in the regulation of auxin biosynthesis by cytokinin. Sci. Rep. 2016, 6, 36866. [Google Scholar] [CrossRef] [PubMed]
- Dłużewska, J.; Szymańska, R.; Gabruk, M.; Kós, P.B.; Nowicka, B.; Kruk, J. Tocopherol Cyclases-Substrate Specificity and Phylogenetic Relations. PLoS ONE 2016, 11, e0159629. [Google Scholar] [CrossRef]
- DellaPenna, D.; Pogson, B.J. Vitamin synthesis in plants: Tocopherols and carotenoids. Annu. Rev. Plant Biol. 2006, 57, 711–738. [Google Scholar] [CrossRef]
- Nagegowda, D.A.; Gupta, P. Advances in biosynthesis, regulation, and metabolic engineering of plant specialized terpenoids. Plant Sci. 2020, 294, 110457. [Google Scholar] [CrossRef]
- Tian, S.; Wang, D.; Yang, L.; Zhang, Z.; Liu, Y. A systematic review of 1-Deoxy-D-xylulose-5-phosphate synthase in terpenoid biosynthesis in plants. Plant Growth Regul. 2022, 96, 221–235. [Google Scholar] [CrossRef]
- Xie, Z.; Nolan, T.M.; Jiang, H.; Yin, Y. AP2/ERF Transcription Factor Regulatory Networks in Hormone and Abiotic Stress Responses in Arabidopsis. Front. Plant Sci. 2019, 10, 228. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Sample | Total Clean Reads | Total Clean Bases | Q30 Percentage (%) | GC Percentage (%) | Mapped (%) | Sequence Depth |
|---|---|---|---|---|---|---|
| Female | 58,761,436 | 17,593,008,450 | 93.87 | 37.51 | 93.28 | 23 |
| Male | 67,321,840 | 20,156,470,118 | 93.53 | 37.70 | 90.83 | 25 |
| Offspring | 2,559,946,028 | 765,858,305,730 | 92.78 | 37.51 | 94.86 | 4.26 |
| Total | 2,686,029,304 | 803,607,784,298 |
| LG ID | Marker Number | Total Distance (cM) | Average Distance (cM) | Max Gap (cM) |
|---|---|---|---|---|
| LG01 | 470 | 144.97 | 0.31 | 1.85 |
| LG02 | 267 | 95.07 | 0.36 | 1.85 |
| LG03 | 320 | 88.96 | 0.28 | 1.38 |
| LG04 | 346 | 102.23 | 0.30 | 1.85 |
| LG05 | 221 | 62.26 | 0.28 | 1.15 |
| LG06 | 260 | 103.63 | 0.40 | 5.02 |
| LG07 | 208 | 99.03 | 0.48 | 2.56 |
| LG08 | 313 | 108.45 | 0.35 | 6.24 |
| LG09 | 140 | 45.51 | 0.33 | 2.56 |
| LG10 | 318 | 143.22 | 0.45 | 7.86 |
| LG11 | 248 | 73.22 | 0.30 | 0.92 |
| LG12 | 180 | 50.63 | 0.28 | 1.15 |
| LG13 | 258 | 81.74 | 0.32 | 1.38 |
| LG14 | 187 | 55.68 | 0.30 | 1.62 |
| LG15 | 258 | 82.28 | 0.32 | 2.8 |
| LG16 | 274 | 91.64 | 0.34 | 4.52 |
| Total | 4268 | 1428.51 | 0.33 |
| Traits | QTL Name | LOD Threshold | Linkage Group | Position (cM) | PVE (%) |
|---|---|---|---|---|---|
| Shikimic acid content | qSK-12.1 | 3 | 12 | 12.543–13.228 | 7.5 |
| qSK-12.2 | 3 | 12 | 13.912–25.995 | 8.2 |
| Species | Number of Individuals | Number of Makers | Length (cM) | Density (cM) | Traits of QTL | Reference |
|---|---|---|---|---|---|---|
| Populus | 500 | 5796 | 2683.80 | 0.46 | Leaf related traits | [28] |
| Ziziphus jujuba Mill. | 140 | 8684 | 1713.22 | 0.2 | Leaf related traits | [50] |
| Lycium Linn. | 200 | 8507 | 2122.24 | 0.25 | Leaf and fruit related traits | [49] |
| Camellia sinensis | 96 | 8596 | 1490.81 | 0.166 | Leaf area | [32] |
| Camellia sinensis | 58 | 5250 | 3328.51 | 0.68 | Zigzag-shaped stem | [51] |
| Eriobotrya japonica Lindl. | 130 | 3859 | 1988.12 | 0.52 | Fruit weight | [30] |
| peanut | 200 | 2802 | 1573.85 | 0.58 | Bacterial resistance | [52] |
| Grapes | 105 | 1662 | 1463.38 | 0.88 | Firmness | [53] |
| eggplant | 200 | 3776 | 2022.8 | 0.56 | Color of leaf and fruit | [54] |
| This study | 220 | 4268 | 1428.51 | 0.33 | shikimic acid content |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Li, H.; Li, Y.; Bao, F.; Zhan, D.; Pang, Z.; Zhao, J.; Zhang, J. Construction of the First High-Density Genetic Linkage Map and QTL Mapping of Shikimic Acid Content in Liquidambar. Forests 2024, 15, 1662. https://doi.org/10.3390/f15091662
Fan Y, Li H, Li Y, Bao F, Zhan D, Pang Z, Zhao J, Zhang J. Construction of the First High-Density Genetic Linkage Map and QTL Mapping of Shikimic Acid Content in Liquidambar. Forests. 2024; 15(9):1662. https://doi.org/10.3390/f15091662
Chicago/Turabian StyleFan, Yingming, Hongxuan Li, Ying Li, Fen Bao, Dingju Zhan, Zhenwu Pang, Jian Zhao, and Jinfeng Zhang. 2024. "Construction of the First High-Density Genetic Linkage Map and QTL Mapping of Shikimic Acid Content in Liquidambar" Forests 15, no. 9: 1662. https://doi.org/10.3390/f15091662