De Novo Transcriptomic Analysis and Development of EST–SSRs for Styrax japonicus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA and RNA Isolation
2.3. cDNA Library Construction and Sequencing
2.4. Sequence Assembly and Annotation
2.5. Identification of EST–SSR Loci and Primer Design
2.6. Validation and Application of SSR Markers
3. Results
3.1. Sequencing and De Novo Assembly
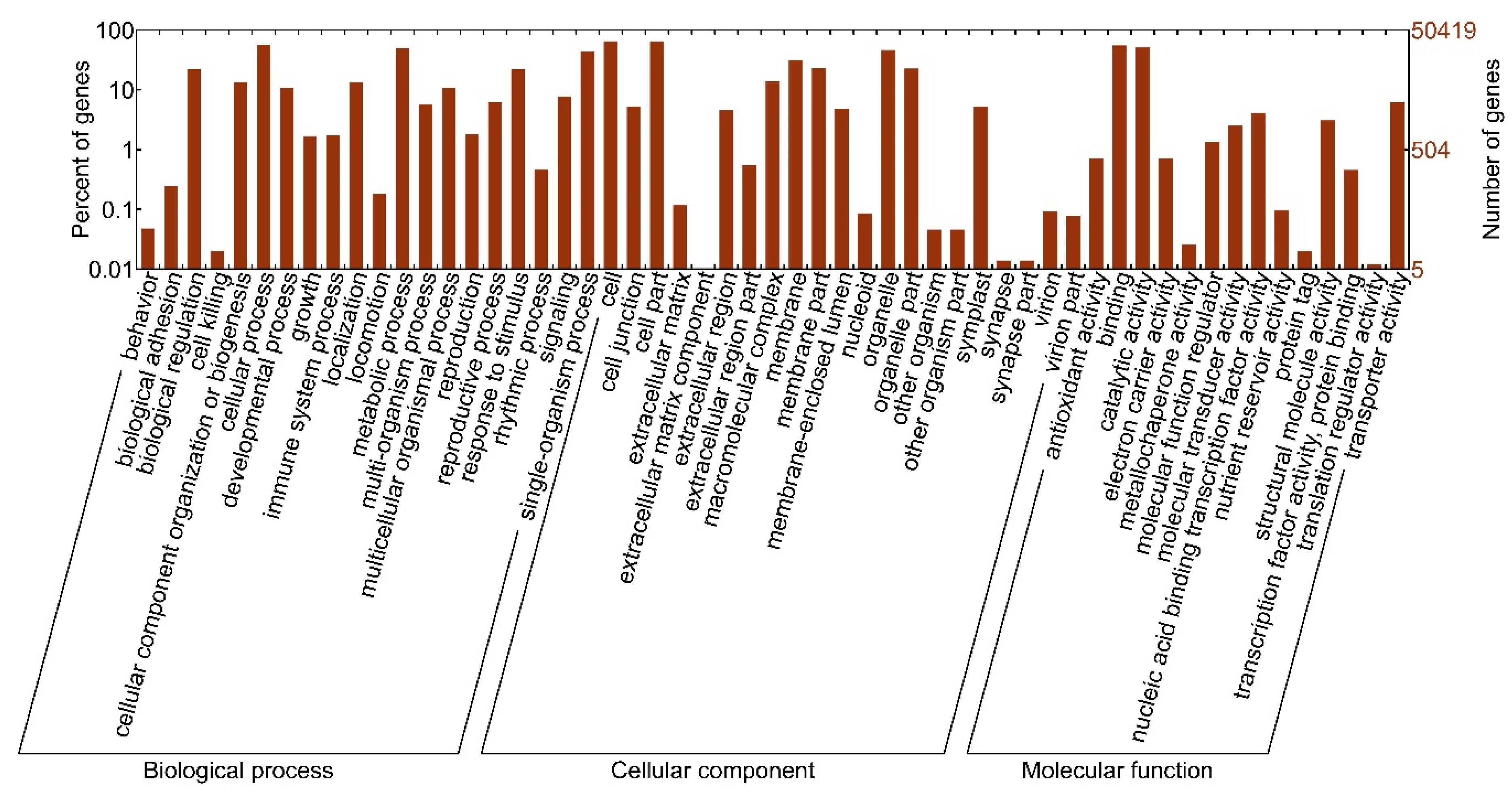
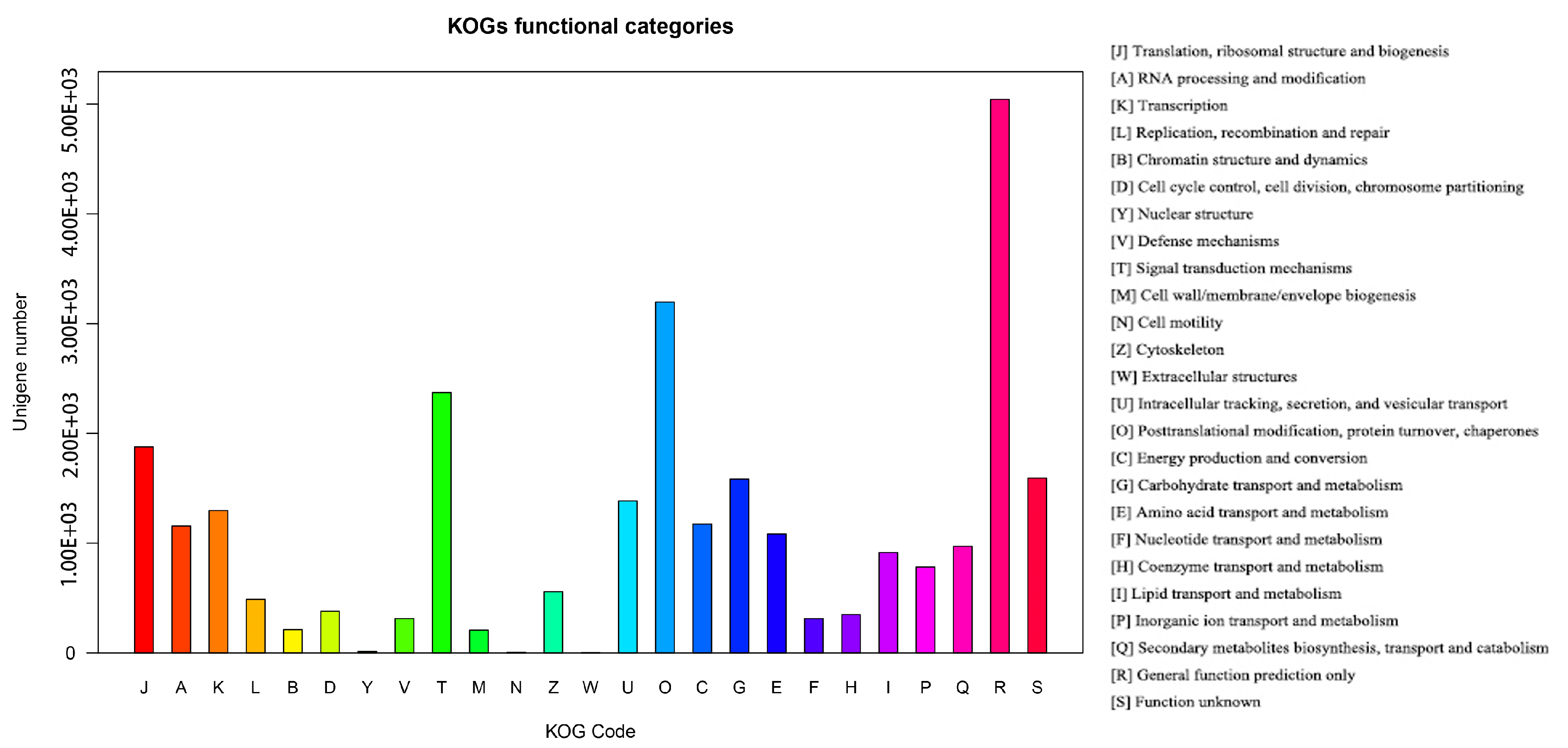
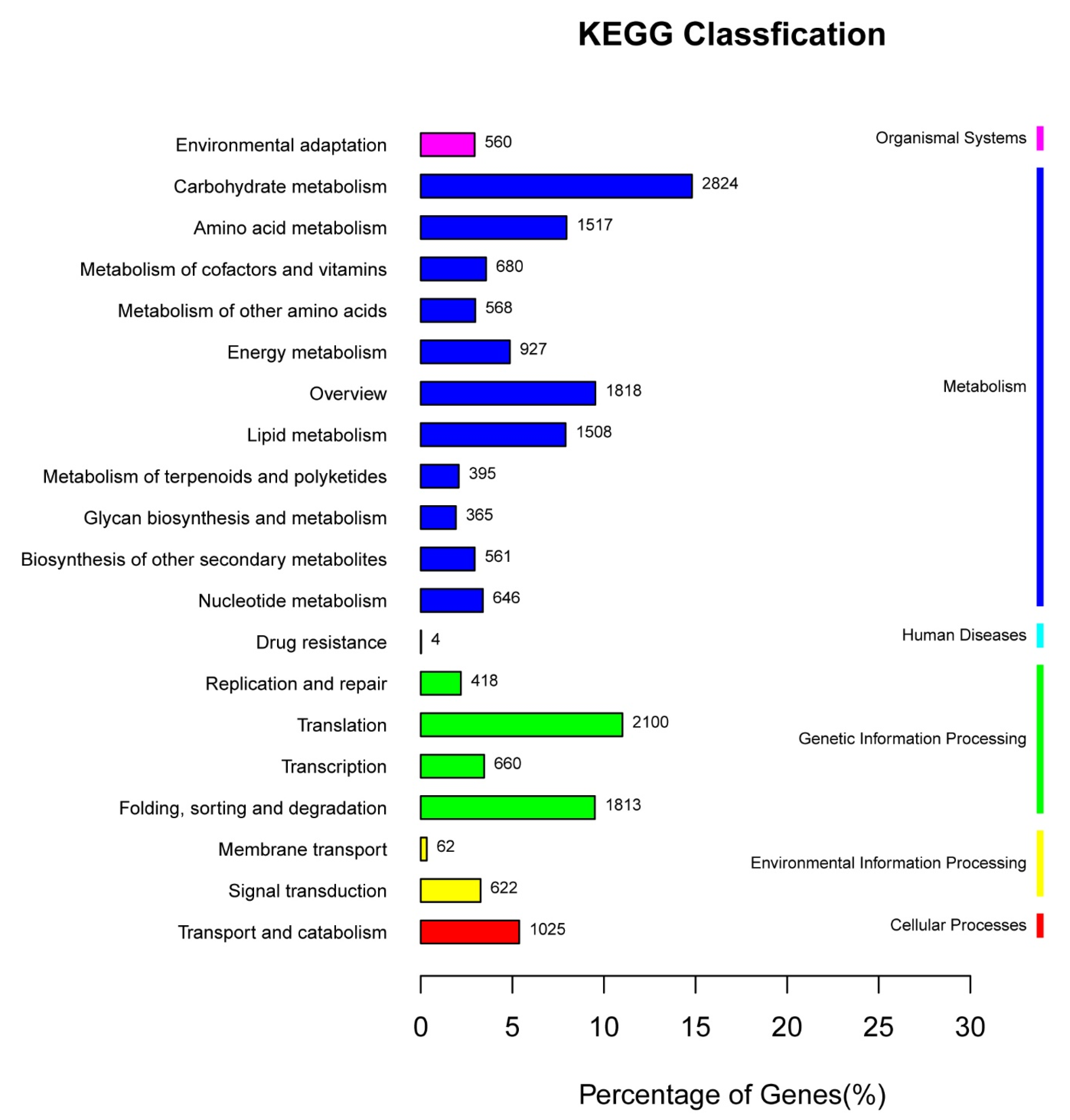
3.2. Functional Annotation of Unigenes
3.3. Frequency and Distribution of SSRs
3.4. Development and Validation of Novel EST–SSRs
4. Discussion
4.1. Characterization of the S. japonicus Transcriptome
4.2. SSR Markers in the Transcriptome of S. japonicus
4.3. Validation of EST-SSR Markers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, X.Y.; Yu, S.; Liu, M.; Yang, Q.S.; Chen, X.Y. Twenty-three microsatellite loci for Styrax confusus and Styrax japonicus (Styracaceae). Conserv. Genet. Resour. 2009, 2, 51–54. [Google Scholar] [CrossRef]
- Wang, L.; Wang, K.L.; Liu, Q.C.; Liu, Q.H. Effects of gibberellin and lamination treatment on seed germination of Styrax japonicus. Acta. Agric. Jiangxi. 2010, 22, 77–79. [Google Scholar]
- Horimoto, T.; Koshioka, M.; Kubota, S.; Mander, L.N.; Hirai, N.; Ishida, N.; Suh, J.K.; Lee, A.K.; Roh, M.S. Effect of warm and cold stratification on 1 H-NMR profiles, endogenous gibberellins and abscisic acid in Styrax japonicus seeds. Hortic. Environ. Biote. 2011, 52, 233. [Google Scholar] [CrossRef]
- Shim, K.K.; Ha, Y.M. Mass propagation of Korean Native Styrax japonicus through axillary bud culture. J. Korean Soc. Hortic. Sci. 1997, 38, 575–580. [Google Scholar]
- Ismail, N.A.; Rafii, M.Y.; Mahmud, T.M.; Hanafi, M.M.; Miah, G. Molecular markers: A potential resource for ginger genetic diversity studies. Mol. Biol. Rep. 2016, 43, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Oh, E.U.; Min, S.L.; Kim, H.B.; Moon, D.G.; Song, K.J. Construction of a genetic linkage map based on RAPD, AFLP, and SSR markers for tea plant (Camellia sinensis). Euphytica 2017, 213, 190. [Google Scholar] [CrossRef]
- Phumichai, C.; Phumichai, T.; Wongkaew, A. Novel chloroplast microsatellite (cpSSR) markers for genetic diversity assessment of cultivated and wild Hevea Rubber. Plant Mol. Biol. Rep. 2015, 33, 1–13. [Google Scholar] [CrossRef]
- Zhou, Q.; Luo, D.; Ma, L.; Xie, W.; Wang, Y.; Wang, Y.; Liu, Z. Development and cross-species transferability of EST-SSR markers in Siberian wildrye (Elymus sibiricus L.) using Illumina sequencing. Sci. Rep. 2016, 6, 20549. [Google Scholar] [CrossRef] [PubMed]
- Yoichi, W.; Sakaguchi, S.; Ueno, S.; Tomaru, N.; Uehara, K. Development and characterization of EST-SSR markers for the genus Rhododendron section Brachycalyx (Ericaceae). Plant Spec. Biol. 2017, 32, 455–459. [Google Scholar] [CrossRef]
- Zheng, X.; Cheng, P.; Ying, D.; You, Y.; Yang, C.; Hu, Z. Development of microsatellite markers by transcriptome sequencing in two species of Amorphophallus (Araceae). BMC Genom. 2013, 14, 490. [Google Scholar] [CrossRef] [PubMed]
- Najafzadeh, R.; Arzani, K.; Bouzari, N.; Saei, A. Genetic variation and identification of promising sour cherries inferred from microsatellite markers. Russ. J. Genet. 2016, 52, 64–73. [Google Scholar] [CrossRef]
- Liu, Y.C.; Liu, S.; Liu, D.C.; Wei, Y.X.; Liu, C.; Yang, Y.M.; Tao, C.G.; Liu, W.S. Exploiting EST databases for the development and characterization of EST-SSR markers in blueberry (Vaccinium) and their cross-species transferability in Vaccinium spp. Sci. Hortic. 2014, 176, 319–329. [Google Scholar] [CrossRef]
- Chen, L.Y.; Cao, Y.N.; Yuan, N.; Nakamura, K.; Wang, G.M.; Qiu, Y.X. Characterization of transcriptome and development of novel EST-SSR makers based on next-generation sequencing technology in Neolitsea sericea (Lauraceae) endemic to East Asian land-bridge islands. Mol. Breed. 2015, 35, 187. [Google Scholar] [CrossRef]
- Taheri, S.; Abdullah, T.L.; Jain, S.M.; Sahebi, M.; Azizi, P. TILLING, high-resolution melting (HRM), and next-generation sequencing (NGS) techniques in plant mutation breeding. Mol. Breed. 2017, 37, 40. [Google Scholar] [CrossRef]
- Ekblom, R.; Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Wang, K.; Su, S.; Tian, R.; Li, Y.; Chen, M. De novo transcriptome analysis and microsatellite marker development for population genetic study of a serious insect pest, Rhopalosiphum padi (L.) (Hemiptera: Aphididae). PLoS ONE 2017, 12, e0172513. [Google Scholar] [CrossRef] [PubMed]
- Severin, A.J.S.; Woody, J.L.; Bolon, Y.T.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Zenoni, S.; Delledonne, M. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 2010, 152, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Squirrell, J.; Hollingsworth, P.M.; Woodhead, M.; Russell, J.; Lowe, A.J.; Gibby, M.; Powell, W. How much effort is required to isolate nuclear microsatellites from plants? Mol. Ecol. 2003, 12, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.C.; Yavuz, A.; Thygesen, H.H.; Erno, V.; Vossen, R.H.A.M.; De, M.R.X.; Boer, J.M.; Van, O.G.J.B.; Den, D.J.T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008, 36, e141. [Google Scholar]
- Liu, C.; Dou, Y.; Guan, X.; Fu, Q.; Zhang, Z.; Hu, Z.; Zheng, J.; Lu, Y.; Li, W. De novo transcriptomic analysis and development of EST-SSRs for Sorbus pohuashanensis (Hance) Hedl. PLoS ONE 2017, 12, e0179219. [Google Scholar] [CrossRef] [PubMed]
- Kovi, M.R.; Amdahl, H.; Alsheikh, M.; Rognli, O.A. De novo and reference transcriptome assembly of transcripts expressed during flowering provide insight into seed setting in tetraploid red clover. Sci. Rep. 2017, 7, 44383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatanparast, M.; Shetty, P.; Chopra, R.; Doyle, J.J.; Sathyanarayana, N.; Egan, A.N. Transcriptome sequencing and marker development in winged bean (Psophocarpus tetragonolobus; Leguminosae). Sci. Rep. 2016, 6, 29070. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, U.K.; Pruthi, V.; Randhawa, G.S. RNA-Seq of Guar (Cyamopsis tetragonoloba, L. Taub.) Leaves: De novo transcriptome assembly, functional annotation and development of genomic resources. Front Plant Sci. 2017, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Wang, Y.H.; Shen, S.K. De dovo assembly of transcriptome and development of novel EST-SSR markers in Rhododendron rex through Illumina sequencing. Front Plant Sci. 2017, 8, 1664. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Deng, M.; Zheng, S.S.; Song, Y.G. De novo transcriptome assembly and development of SSR markers of oaks Quercus austrocochinchinensis and Q. kerrii (Fagaceae). Tree Gene Genomes. 2016, 12, 103. [Google Scholar] [CrossRef]
- Li, J.; Wang, S.; Jing, Y.; Ling, W. A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 2013, 48, 72–78. [Google Scholar]
- Ghawana, S.; Paul, A.; Kumar, H.; Kumar, A.; Singh, H.; Bhardwaj, P.K.; Rani, A.; Singh, R.S.; Raizada, J.; Singh, K. An RNA isolation system for plant tissues rich in secondary metabolites. BMC Res. Notes 2011, 4, 85. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; Garcíagómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J. DNA protocols for plants-CTAB total DNA isolation. Mol. Tech. Taxon. 1991, 283–293. [Google Scholar] [CrossRef]
- Liu, W.; Li, R.; Ayalew, H.; Xia, Y.; Bai, G.; Yan, G.; Siddique, K.H.; Guo, P. Development of a simple and effective silver staining protocol for detection of DNA fragments. Electrophoresis 2017, 38, 1175. [Google Scholar] [CrossRef] [PubMed]
- Krawczak, M.; Nikolaus, S.; Von, E.H.; Croucher, P.J.P.; El, M.N.E.; Schreiber, S. PopGen: Population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Commun. Genet. 2006, 9, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Nagy, S.; Poczai, P.; Cernák, I.; Gorji, A.M.; Hegedűs, G.; Taller, J. PICcalc: An online program to calculate polymorphic information content for molecular genetic studies. Biochem. Genet. 2012, 50, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Zargar, S.M.; Raatz, B.; Sonah, H.; MuslimaNazir; Bhat, J.A.; Dar, Z.A.; Agrawal, G.K.; Rakwal, R. Recent advances in molecular marker techniques: Insight into QTL mapping, GWAS and genomic selection in plants. J. Crop Sci. Biotechnol. 2015, 18, 293–308. [Google Scholar] [CrossRef]
- Carneiro, V.M.L.; Luciane, S.; Lima, D.A.; Freitas, M.C.D. Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biol. 2016, 39, 312–328. [Google Scholar]
- Dang, M.; Liu, Z.X.; Chen, X.; Zhang, T.; Zhou, H.J.; Hu, Y.H.; Zhao, P. Identification, development, and application of 12 polymorphic EST-SSR markers for an endemic Chinese walnut (Juglans cathayensis L.) using next-generation sequencing technology. Biochem. Syst. Ecol. 2015, 60, 74–80. [Google Scholar] [CrossRef]
- Tian, W.; Paudel, D.; Vendrame, W.; Wang, J. Enriching genomic resources and marker development from transcript sequences of Jatropha curcas for Microgravity Studies. Int. J. Genomics 2017, 1, 8614160. [Google Scholar]
- Jia, H.; Yang, H.; Sun, P.; Li, J.; Zhang, J.; Guo, Y.; Han, X.; Zhang, G.; Lu, M.; Hu, J. De novo transcriptome assembly, development of EST-SSR markers and population genetic analyses for the desert biomass willow, Salix psammophila. Sci. Rep. 2016, 6, 39591. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Li, B.; Lin, F.; Huang, P.; Guo, W.; Zheng, Y. De novo sequencing and assembly analysis of transcriptome in Pinus bungeana Zucc. ex Endl. Forests. 2018, 9, 156. [Google Scholar] [CrossRef]
- Xing, W.; Liao, J.; Cai, M.; Xia, Q.; Liu, Y.; Zeng, W.; Jin, X. De novo assembly of transcriptome from Rhododendron latoucheae Franch. using Illumina sequencing and development of new EST-SSR markers for genetic diversity analysis in Rhododendron. Tree Genet. Genomes 2017, 13, 53. [Google Scholar] [CrossRef]
- Feng, C.; Chen, M.; Xu, C.J.; Bai, L.; Yin, X.R.; Li, X.; Allan, A.C.; Ferguson, I.B.; Chen, K.S. Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genom. 2012, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, D.; Li, J.; Yu, B.; Qiao, X.; Huang, H.; He, Y. De novo characterization of leaf transcriptome using 454 sequencing and development of est-ssr markers in Tea (Camellia sinensis). Plant Mol. Biol. Rep. 2013, 31, 524–538. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Huang, X.; Li, J.; Wang, H.; Li, J. De novo assembly and characterization of fruit transcriptome in Litchi chinensis Sonn and analysis of differentially regulated genes in fruit in response to shading. BMC Genom. 2013, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Deng, Z.; Qin, B.; Liu, X.; Men, Z. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genom. 2012, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, L.; Wang, S.; Liu, C.; Blair, M.W.; Cheng, X. Transcriptome sequencing of mung bean (Vigna radiate L.) genes and the identification of EST-SSR markers. PLoS ONE 2015, 10, e0120273. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Feuerstein, U.; Luesink, W.; Schulze, S.; Asp, T.; Studer, B.; Becker, H.C.; Dehmer, K.J. DArT, SNP, and SSR analyses of genetic diversity in Lolium perenne L. using bulk sampling. BMC Genet. 2018, 19, 10. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.V.; Poornima, H.S.; Sivaprasad, V.; Naik, V.G. A new set of mulberry-specific SSR markers for application in cultivar identification and DUS testing. J. Genet. 2018, 97, 1–7. [Google Scholar] [CrossRef]
- Marconi, T.G.; Costa, E.A.; Miranda, H.R.; Mancini, M.C.; Cardoso-Silva, C.B.; Oliveira, K.M. Functional markers for gene mapping and genetic diversity studies in sugarcane. BMC Res. Notes 2011, 4, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.P.; Liu, C.L.; Wu, D.J.; Li, L.; Shu, J.; Sun, C.; Xia, Y.; Zhao, L.J. De novo transcriptome analysis of Fraxinus velutina using Illumina platform and development of EST-SSR markers. Biol. Plantarum. 2016, 61, 210–218. [Google Scholar] [CrossRef]
- Jia, X.; Deng, Y.; Sun, X.; Liang, L.; Su, J. De novo assembly of the transcriptome of Neottopteris nidus using Illumina paired-end sequencing and development of EST-SSR markers. Mol. Breed. 2016, 36, 94. [Google Scholar] [CrossRef]
- Morgante, M.; Hanafey, M.; Powell, W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002, 30, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Cavagnaro, P.F.; Senalik, D.A.; Yang, L.; Simon, P.W.; Harkins, T.T.; Kodira, C.D.; Huang, S.; Weng, Y. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom. 2010, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cai, C.; Cheng, F.; Cui, H.; Zhou, H. Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol. Breed. 2014, 34, 1853–1866. [Google Scholar] [CrossRef]
- Biswas, M.K.; Chai, L.; Mayer, C.; Xu, Q.; Guo, W.; Deng, X. Exploiting BAC-end sequences for the mining, characterization and utility of new short sequences repeat (SSR) markers in Citrus. Mol. Biol. Rep. 2012, 39, 5373–5386. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhang, T.; Gao, X.X.; Wang, Y.; Zhang, Q.; Zhou, H.J. De novo assembly and characterization of the leaf, bud, and fruit transcriptome from the vulnerable tree Juglans mandshurica for the development of 20 new microsatellite markers using Illumina sequencing. Mol. Genet. Genom. 2016, 291, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Items | Number |
|---|---|---|
| Raw reads | Total raw reads | 364,794,740 |
| Clean reads | Total clean reads | 338,570,222 |
| Total clean nucleotide (nt) | 49,540,935,462 | |
| Q20 percentage (average) | 98.01% | |
| Q30 percentage (average) | 94.67% | |
| GC percentage (average) | 41.58% | |
| Unigenes | Total sequence number | 136,071 |
| Total sequence Length | 83,181,839 | |
| Largest (bp) | 10,742 | |
| Smallest (bp) | 224 | |
| Average (bp) | 611 | |
| N50 (bp) | 846 | |
| EST–SSRs | Sequences examined | 136,071 |
| Size of examined sequences (bp) | 83,181,839 | |
| Identified SSRs | 55,977 | |
| SSR-containing sequences | 38,611 | |
| Sequences containing more than one SSR | 11,652 | |
| SSRs present in compound formation | 8121 |
| Repeats | Di- | Tri- | Tetra- | Penta- | Hexa- | Total | Percentage (%) |
|---|---|---|---|---|---|---|---|
| 3 | 5978 | 2170 | 2291 | 10,439 | 25.14 | ||
| 4 | 7072 | 974 | 390 | 624 | 9,060 | 21.82 | |
| 5 | 7064 | 2256 | 262 | 91 | 66 | 9,739 | 23.45 |
| 6 | 2826 | 909 | 80 | 6 | 51 | 3,872 | 9.32 |
| 7 | 1747 | 438 | 11 | 4 | 27 | 2,227 | 5.36 |
| 8 | 1486 | 308 | 8 | 3 | 12 | 1,817 | 4.38 |
| 9 | 1076 | 51 | 4 | 9 | 1,140 | 2.75 | |
| 10 | 824 | 63 | 4 | 3 | 894 | 2.15 | |
| 11 | 687 | 44 | 1 | 3 | 735 | 1.77 | |
| 12 | 453 | 32 | 1 | 486 | 1.17 | ||
| 13 | 130 | 31 | 1 | 162 | 0.39 | ||
| 14 | 139 | 9 | 1 | 149 | 0.36 | ||
| 15 | 147 | 12 | 159 | 0.38 | |||
| 16 | 102 | 6 | 108 | 0.26 | |||
| 17 | 93 | 7 | 100 | 0.24 | |||
| 18 | 87 | 3 | 90 | 0.22 | |||
| 19 | 68 | 7 | 75 | 0.18 | |||
| 20 | 55 | 3 | 58 | 0.14 | |||
| 21 | 41 | 3 | 44 | 0.11 | |||
| 22 | 31 | 2 | 33 | 0.08 | |||
| 23 | 16 | 1 | 17 | 0.04 | |||
| ≥24 | 121 | 3 | 2 | 126 | 0.30 | ||
| Total | 17,193 | 11,260 | 7323 | 2664 | 3090 | 41,530 | |
| Percentage (%) | 41.40 | 27.11 | 17.64 | 6.41 | 7.44 |
| Primer | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Tm (°C) | SSRs | Alleles Number | Ho | He | PIC | Fst | r |
|---|---|---|---|---|---|---|---|---|---|---|
| Sty-3 | GGATCGATCCCAATGGCGTA | TCCTTAGCTCCATCAAGTGCA | 59.091 | (GTTTG) 5 | 7 | 0.800 | 0.625 | 0.582 | 0.023 | 0.143 |
| Sty-7 | ACAAGCCATGAGCCGATGAA | TGTTGTCCTTTGTTAATTCCCCT | 58.19 | (AGC) 4 | 3 | 0.325 | 0.308 | 0.282 | 0.049 | 0.333 |
| Sty-16 | CTCTCCACCCCAATTGCCAT | CCAAGCTTGCCAAATCCACC | 60.037 | (CCG) 8 | 7 | 0.475 | 0.464 | 0.441 | 0.037 | 0.143 |
| Sty-20 | GGGTTGTATAAGGGTTGCCA | GACGGTGGTACTTCACAGTGT | 59.932 | (AT) 10 | 7 | 0.692 | 0.767 | 0.732 | 0.019 | 0.143 |
| Sty-39 | TTCATCGGCTCTTGGCTTGT | CAGCGTCAAGTTGGTTTCGG | 60.041 | (GTG) 4 | 9 | 0.950 | 0.811 | 0.776 | 0.027 | 0.111 |
| Sty-43 | GGACGCTCAAGGTCAACTCA | AGGCTGCCATATCTAGGGGT | 59.805 | (TCGGT) 3 | 5 | 0.875 | 0.603 | 0.523 | 0.005 | 0.200 |
| Sty-44 | CCTTCCCCACAACCCACTAC | ATGCTTCCTCATTGTGCAGC | 59.184 | (TC) 12 | 12 | 0.939 | 0.882 | 0.855 | 0.039 | 0.083 |
| Sty-47 | GGGTTGGCCACCTTGATCTT | TCAGAGGCTCCAATACGTAGC | 59.318 | (TC) 11 (TA) 8 | 14 | 1.000 | 0.873 | 0.848 | 0.073 | 0.071 |
| Sty-49 | TCACCAGTTCGCGTGTTAGT | GCATGGGGTTTCTTGCGAAG | 60.109 | (TCAT) 3 | 6 | 0.846 | 0.725 | 0.678 | 0.027 | 0.167 |
| Sty-55 | TGTTGCTTGAAAGTGCGGTC | GTGTGGACACATCACCGGAT | 60.036 | (AATA) 4 | 8 | 0.256 | 0.491 | 0.450 | 0.043 | 0.125 |
| Sty-57 | TCGGGATCTTTTAAGTTTTGGAGC | GAATCCGTTTGGTTGCCGAG | 59.832 | (AAAT) 3 | 3 | 0.025 | 0.074 | 0.071 | 0.030 | 0.333 |
| Sty-58 | TCCTTGTGCACTCAGTCCAC | TGTAGCACACGGTCCTGTTC | 59.967 | (GGTTC) 3 | 2 | 0.077 | 0.266 | 0.226 | 0.120 | 0.500 |
| Sty-59 | TGCTTCAAAATGCATTGCCA | GGATCCACAGTTGCATCAGC | 59.263 | (GAAT) 3 | 3 | 0.900 | 0.553 | 0.442 | 0.012 | 0.333 |
| Sty-61 | ACTTGGCTAGAGAGGGGGAG | AGGTGAGTGAGAATTGGGCG | 60.036 | (CGC) 4 | 2 | 0.550 | 0.404 | 0.319 | 0.154 | 0.500 |
| Sty-66 | CGGTGTTGAGAGGAGTCGTC | CCAGCATCTCCTCCTTCCAG | 59.531 | (AGT) 5 | 4 | 0.316 | 0.413 | 0.374 | 0.041 | 0.250 |
| Sty-69 | CCACTCGAGCCAACCAACAT | AGACGAGACTGAGTGGAGTT | 57.72 | (TC) 5 | 2 | 0.400 | 0.324 | 0.269 | 0.004 | 0.500 |
| Sty-72 | GTGACAGAGGGGAGCACTTC | CCGCCCAATGTAGTGAAGGT | 60.035 | (GTAGGA) 3 | 5 | 0.950 | 0.574 | 0.474 | 0.004 | 0.200 |
| Sty-77 | AAGAGAGGAACAGTGGCAGC | TCGTGTCTACCTCCAACAAAGT | 59.303 | (AG) 11 | 5 | 0.450 | 0.704 | 0.636 | 0.026 | 0.200 |
| Sty-86 | GCAATCGAGAGGGAGTGAGG | CGGTTGGCCAGTTTCTTTGG | 59.968 | (AG) 5 | 2 | 0.375 | 0.309 | 0.258 | 0.173 | 0.500 |
| Sty-87 | GGGCTGGGTCAGAATGTGAA | CGACAACAGTTGCCACCTTG | 59.971 | (GTT) 5 | 5 | 0.725 | 0.565 | 0.517 | 0.017 | 0.200 |
| Sty-88 | GTCGAATTCCGCAAGCCATC | GGTTCACCGTGATTCACCGA | 60.32 | (AT) 6 | 15 | 0.975 | 0.854 | 0.825 | 0.046 | 0.067 |
| Mean | 59.6042 | 6 | 0.614 | 0.552 | 0.504 | 0.046 | 0.243 | |||
| min | 57.72 | 2 | 0.025 | 0.074 | 0.071 | 0.004 | 0.067 | |||
| max | 60.32 | 15 | 1.000 | 0.882 | 0.855 | 0.173 | 0.500 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Zhang, C.; Jiang, X.; Liu, Q.; Liu, Q.; Wang, K. De Novo Transcriptomic Analysis and Development of EST–SSRs for Styrax japonicus. Forests 2018, 9, 748. https://doi.org/10.3390/f9120748
Li W, Zhang C, Jiang X, Liu Q, Liu Q, Wang K. De Novo Transcriptomic Analysis and Development of EST–SSRs for Styrax japonicus. Forests. 2018; 9(12):748. https://doi.org/10.3390/f9120748
Chicago/Turabian StyleLi, Wei, Cuiping Zhang, Xinqiang Jiang, Qingchao Liu, Qinghua Liu, and Kuiling Wang. 2018. "De Novo Transcriptomic Analysis and Development of EST–SSRs for Styrax japonicus" Forests 9, no. 12: 748. https://doi.org/10.3390/f9120748