
The Fatty Acid Lipid Metabolism Nexus in COVID-19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Digestive System Involvement in COVID-19
3. Potential Impact of Elevated Systemic and Cellular Fatty Acid Levels in Obese Individuals Infected with SARS-CoV-2
4. The Essential Role of Lipids during SARS-CoV-2 Replication and Virion Assembly
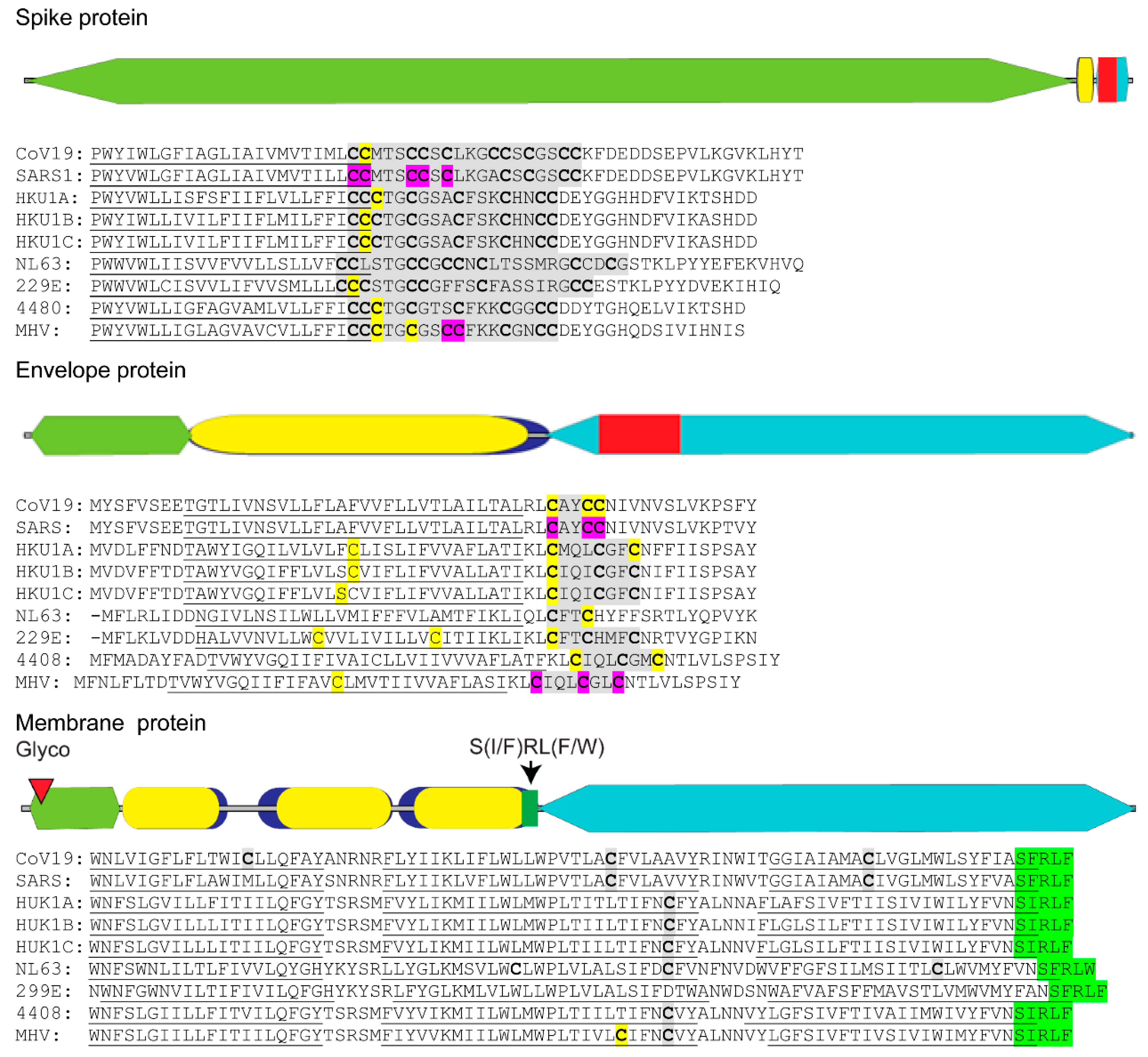
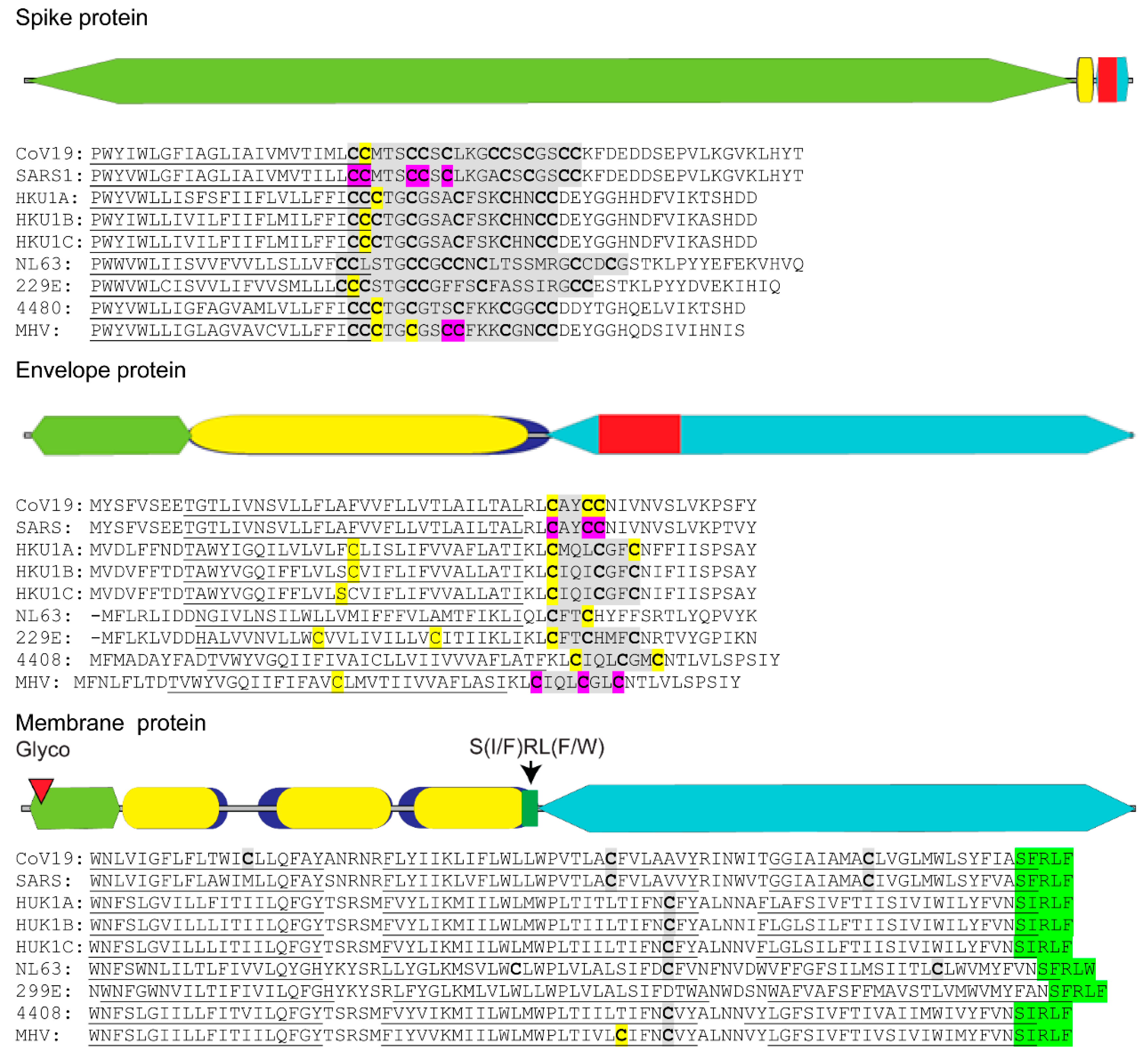
5. The Essential Role of Lipid Addition to SARS-CoV Proteins
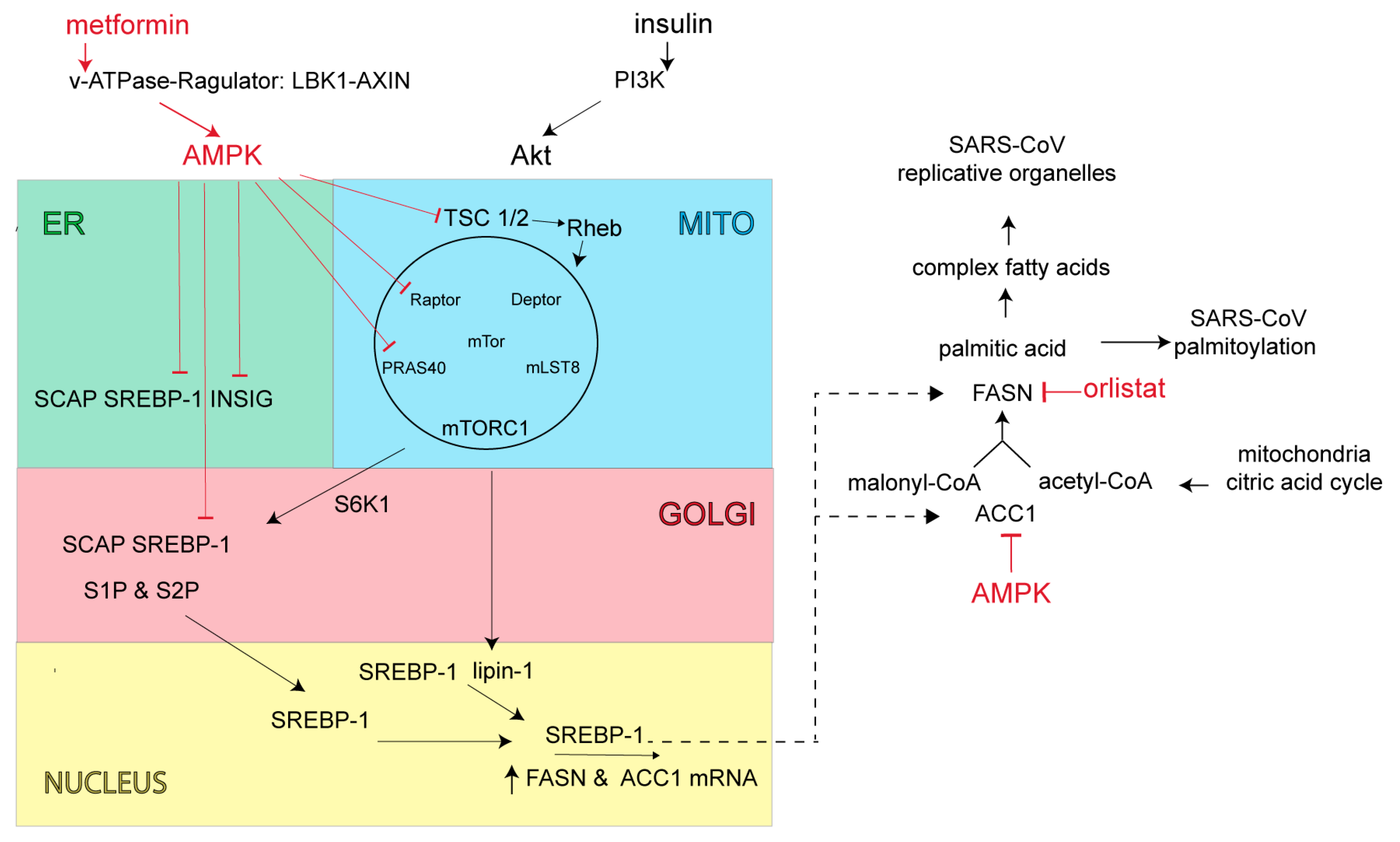
6. Fatty Acid Synthase (FASN) and AMP-Activated Protein Kinase (AMPK): Potential Antiviral Targets to Block SARS-CoV-2 Replication and Virus Assembly
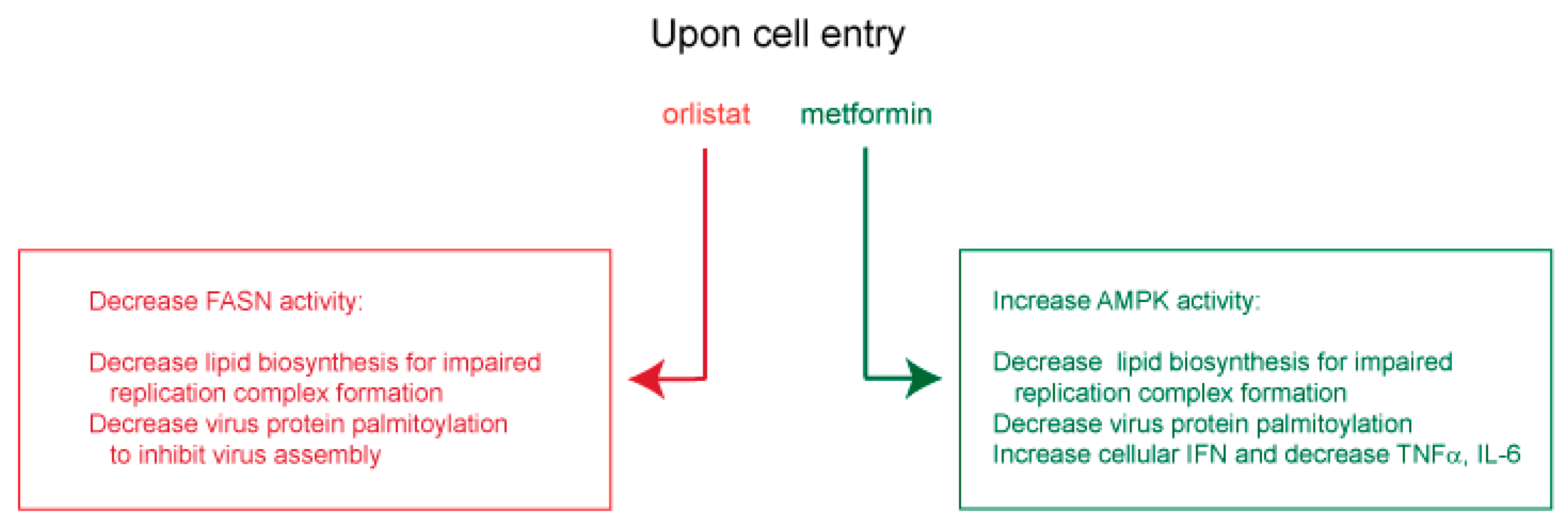
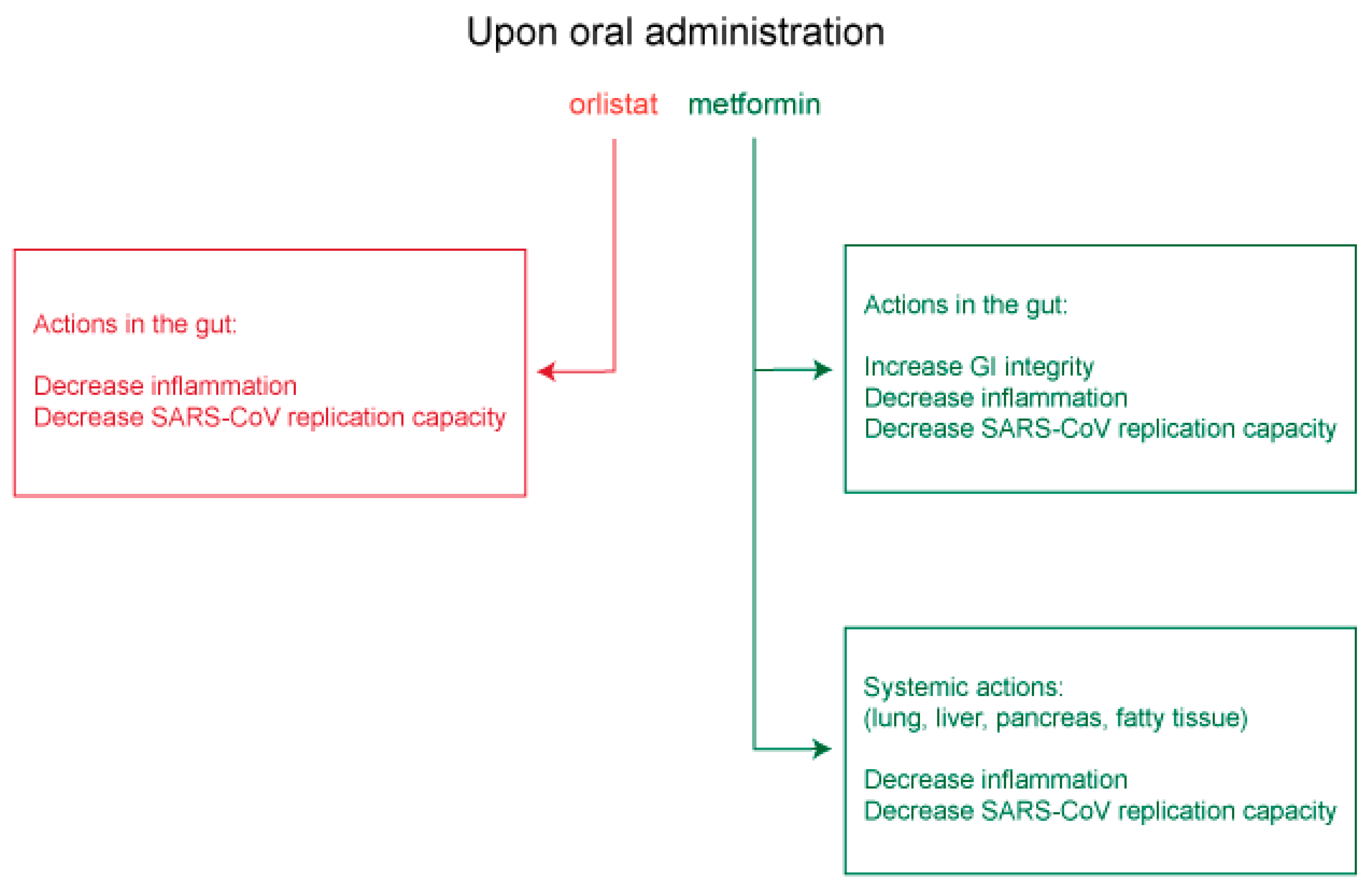
7. Potential Use of Orlistat and Metformin in Controlling SARS-CoV-2 Protein Palmitoylation and Organelle Assembly
7.1. Insights from Other Lipid-Dependent Viruses for Use of Orlistat and Metformin in COVID-19 Treatment
7.2. Potential Clinical Utility of Orlistat and Metformin in COVID-19 Treatment
7.3. Therapeutic Application of Orlistat and Metformin
8. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tontonoz, P. Phospholipid remodeling in physiology and disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Beilstein, F.; Lemasson, M.; Pène, V.; Rainteau, D.; Demignot, S.; Rosenberg, A.R. Lysophosphatidylcholine acyltransferase 1 is downregulated by hepatitis c virus: Impact on production of lipo-viro-particles. Gut 2017, 66, 2160–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.Z. Severe acute respiratory syndrome and its lesions in digestive system. World J. Gastroenterol. 2003, 9, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.M.; Matukas, L.M.; Tomlinson, G.A.; Rachlis, A.R.; Rose, D.B.; Dwosh, H.A.; Walmsley, S.L.; Mazzulli, T.; Avendano, M.; Derkach, P.; et al. Clinical features and short-term outcomes of 144 patients with sars in the greater toronto area. JAMA 2003, 289, 2801–2809. [Google Scholar] [CrossRef] [Green Version]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First case of 2019 novel coronavirus in the united states. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Song, Y.; Liu, P.; Shi, X.L.; Chu, Y.L.; Zhang, J.; Xia, J.; Gao, X.Z.; Qu, T.; Wang, M.Y. Sars-cov-2 induced diarrhoea as onset symptom in patient with covid-19. Gut 2020, 69, 1143–1144. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Jiang, X.; Zhang, Z.; Huang, S.; Zhang, Z.; Fang, Z.; Gu, Z.; Gao, L.; Shi, H.; Mai, L.; et al. Gastrointestinal symptoms of 95 cases with sars-cov-2 infection. Gut 2020, 69, 997–1001. [Google Scholar] [CrossRef]
- Evans, M. The Epidemiology of Severe Acute Respiratory Syndrome: A Global Perspective. In Imaging in SARS; Ahuja, A., Ooi, C., Eds.; Cambridge University Press: Cambridge, UK, 2004; pp. 1–16. [Google Scholar]
- Han, C.; Duan, C.; Zhang, S.; Spiegel, B.; Shi, H.; Wang, W.; Zhang, L.; Lin, R.; Liu, J.; Ding, Z.; et al. Digestive symptoms in covid-19 patients with mild disease severity: Clinical presentation, stool viral rna testing, and outcomes. Am. J. Gastroenterol. 2020, 115, 916–923. [Google Scholar] [CrossRef]
- Jin, X.; Lian, J.S.; Hu, J.H.; Gao, J.; Zheng, L.; Zhang, Y.M.; Hao, S.R.; Jia, H.Y.; Cai, H.; Zhang, X.L.; et al. Epidemiological, clinical and virological characteristics of 74 cases of coronavirus-infected disease 2019 (covid-19) with gastrointestinal symptoms. Gut 2020, 69, 1002–1009. [Google Scholar] [CrossRef] [Green Version]
- Wurtzer, S.; Marechal, V.; Mouchel, J.M.; Maday, Y.; Teyssou, R.; Richard, E.; Almayrac, J.L.; Moulin, L. Evaluation of lockdown effect on SARS-CoV-2 dynamics through viral genome quantification in waste water, Greater Paris, France, 5 March to 23 April 2020. Euro Surveill. 2020, 25, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with sars-cov-2 pneumonia in wuhan, china: A single-centered, retrospective, observational study. Lancet. Respir. Med. 2020, 8, 475–481. [Google Scholar]
- Bornstein, S.R.; Dalan, R.; Hopkins, D.; Mingrone, G.; Boehm, B.O. Endocrine and metabolic link to coronavirus infection. Nat. Rev. Endocrinol. 2020, 16, 297–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Binding of sars coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol. 2010, 47, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, T.N.; Lee, K.C.; Yao, H.; Tsang, T.Y.; Chow, T.C.; Yeung, Y.C.; Choi, K.W.; Tso, Y.K.; Lau, T.; Lai, S.T.; et al. Sars-associated viral hepatitis caused by a novel coronavirus: Report of three cases. Hepatology 2004, 39, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nishida, Y.; Aida, K.; Maruyama, T.; Shimada, A.; Suzuki, M.; Shimura, H.; Takizawa, S.; Takahashi, M.; Akiyama, D.; et al. Enterovirus infection, cxc chemokine ligand 10 (cxcl10), and cxcr3 circuit: A mechanism of accelerated beta-cell failure in fulminant type 1 diabetes. Diabetes 2009, 58, 2285–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vankadari, N.; Wilce, J.A. Emerging wuhan (covid-19) coronavirus: Glycan shield and structure prediction of spike glycoprotein and its interaction with human cd26. Emerg. Microbes Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D., Jr.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. Cd209l (l-sign) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef] [Green Version]
- Petrilli, C.M.; Jones, S.A.; Yang, J.; Rajagopalan, H.; O’Donnell, L.F.; Chernyak, Y.; Tobin, K.; Cerfolio, R.J.; Francois, F.; Horwitz, L.I. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: Prospective cohort study. BMJ. 2020, 369, m1966. [Google Scholar] [CrossRef]
- Genoni, G.; Prodam, F.; Marolda, A.; Giglione, E.; Demarchi, I.; Bellone, S.; Bona, G. Obesity and infection: Two sides of one coin. Eur. J. Pediatrics 2014, 173, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, P. Obesity and respiratory infections: Does excess adiposity weigh down host defense? Pulm. Pharmacol. Ther. 2013, 26, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo, M.E.; Scherer, P.E. Adipose tissue-derived factors: Impact on health and disease. Endocr. Rev. 2006, 27, 762–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiu, J.H.; Dorweiler, B.; Woo, C.W. Interaction between gut microbiota and toll-like receptor: From immunity to metabolism. J. Mol. Med. 2017, 95, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Cnop, M.; Ladriere, L.; Igoillo-Esteve, M.; Moura, R.F.; Cunha, D.A. Causes and cures for endoplasmic reticulum stress in lipotoxic beta-cell dysfunction. DiabetesObes. Metab. 2010, 12 (Suppl. 2), 76–82. [Google Scholar]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar]
- Karagöz, G.E.; Acosta-Alvear, D.; Walter, P. The unfolded protein response: Detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2019, 1, a033886. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tontonoz, P. Liver x receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Altan-Bonnet, N. Lipid tales of viral replication and transmission. Trends Cell Biol. 2017, 27, 201–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, M. Palmitoylation of virus proteins. Biol. Cell 2012, 104, 493–515. [Google Scholar] [CrossRef] [PubMed]
- Stapleford, K.A.; Miller, D.J. Role of cellular lipids in positive-sense rna virus replication complex assembly and function. Viruses 2010, 2, 1055–1068. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Chu, H.; Yang, D.; Sze, K.H.; Lai, P.M.; Yuan, S.; Shuai, H.; Wang, Y.; Kao, R.Y.; Chan, J.F.; et al. Characterization of the lipidomic profile of human coronavirus-infected cells: Implications for lipid metabolism remodeling upon coronavirus replication. Viruses 2019, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Neuman, B.W.; Angelini, M.M.; Buchmeier, M.J. Does form meet function in the coronavirus replicative organelle? Trends Microbiol 2014, 22, 642–647. [Google Scholar] [CrossRef] [Green Version]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Sars-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef]
- de Wilde, A.H.; Snijder, E.J.; Kikkert, M.; van Hemert, M.J. Host factors in coronavirus replication. Curr. Top. Microbiol. Immunol. 2018, 419, 1–42. [Google Scholar]
- Sprooten, J.; Garg, A.D. Type i interferons and endoplasmic reticulum stress in health and disease. Int. Rev. Cell Mol. Biol. 2020, 350, 63–118. [Google Scholar]
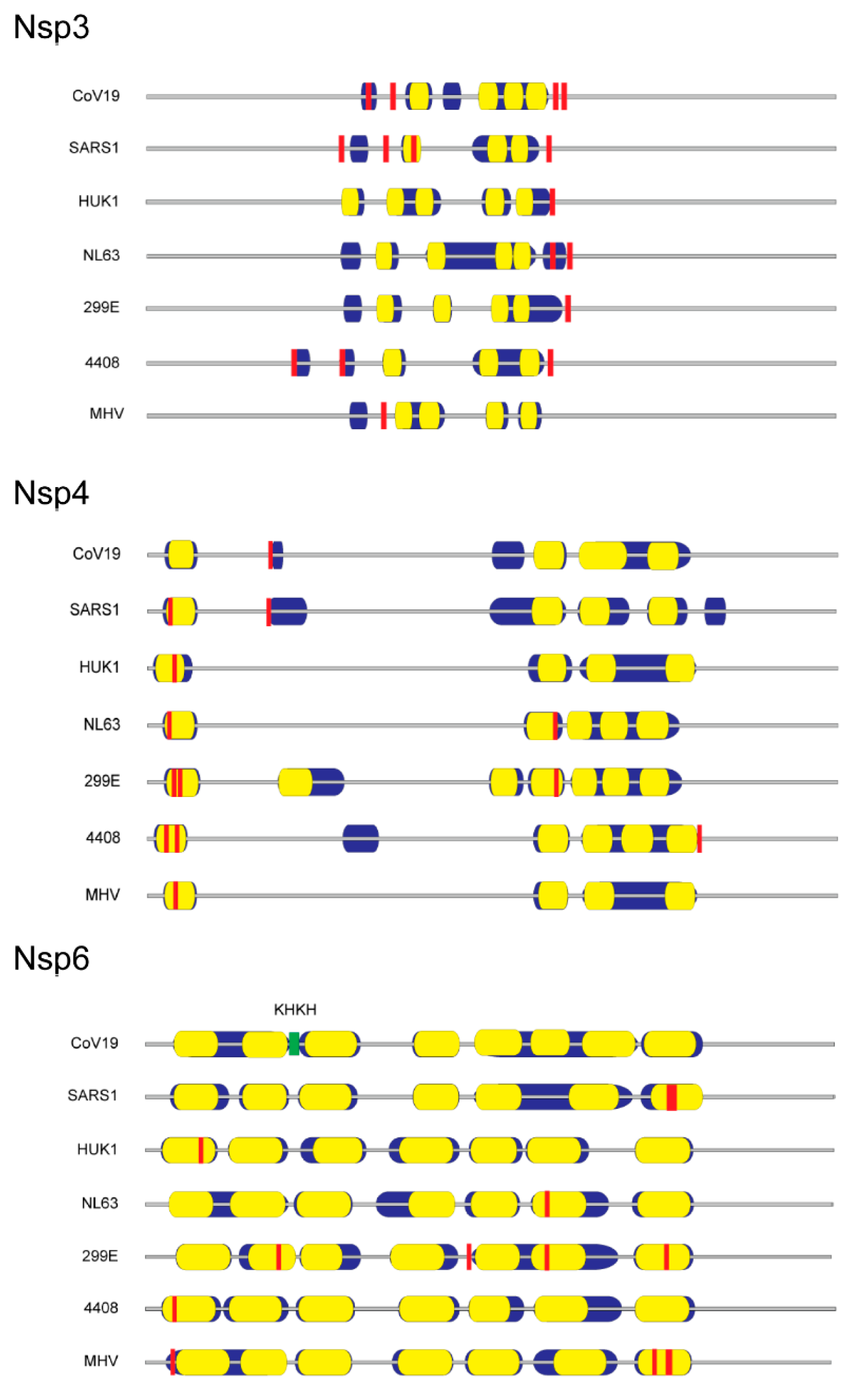
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheije, M.H.; Raaben, M.; Mari, M.; Te Lintelo, E.G.; Reggiori, F.; van Kuppeveld, F.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus rna replication depends on gbf1-mediated arf1 activation. PLoS Pathog. 2008, 4, e1000088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human coronaviruses: A review of virus-host interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kong, L.; Yu, X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit. Rev. Microbiol. 2015, 41, 150–164. [Google Scholar] [CrossRef]
- McBride, C.E.; Machamer, C.E. Palmitoylation of sars-cov s protein is necessary for partitioning into detergent-resistant membranes and cell-cell fusion but not interaction with m protein. Virology 2010, 405, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Boscarino, J.A.; Logan, H.L.; Lacny, J.J.; Gallagher, T.M. Envelope protein palmitoylations are crucial for murine coronavirus assembly. J. Virol. 2008, 82, 2989–2999. [Google Scholar] [CrossRef] [Green Version]
- Thorp, E.B.; Boscarino, J.A.; Logan, H.L.; Goletz, J.T.; Gallagher, T.M. Palmitoylations on murine coronavirus spike proteins are essential for virion assembly and infectivity. J. Virol. 2006, 80, 1280–1289. [Google Scholar] [CrossRef] [Green Version]
- Resh, M.D. Fatty acylation of proteins: The long and the short of it. Prog. Lipid Res. 2016, 63, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, L.H.; Shipston, M.J. The physiology of protein s-acylation. Physiol. Rev. 2015, 95, 341–376. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.G.; Grosenbach, D.W.; Hruby, D.E. Analysis of the site occupancy constraints of primary amino acid sequences in the motif directing palmitylation of the vaccinia virus 37-kda envelope protein. Virology 1999, 254, 124–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, M.S.; Lee, C.J.; Banerjee, A. The molecular mechanism of dhhc protein acyltransferases. Biochem. Soc. Trans. 2019, 47, 157–167. [Google Scholar] [CrossRef]
- Lopez, L.A.; Riffle, A.J.; Pike, S.L.; Gardner, D.; Hogue, B.G. Importance of conserved cysteine residues in the coronavirus envelope protein. J. Virol. 2008, 82, 3000–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, C.M.; Chouljenko, V.N.; Iyer, A.; Colgrove, R.; Farzan, M.; Knipe, D.M.; Kousoulas, K.G. Palmitoylation of the cysteine-rich endodomain of the sars-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology 2007, 360, 264–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Yuan, Q.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology 2006, 349, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Venkatagopalan, P.; Daskalova, S.M.; Lopez, L.A.; Dolezal, K.A.; Hogue, B.G. Coronavirus envelope (e) protein remains at the site of assembly. Virology 2015, 478, 75–85. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef]
- Ruch, T.R.; Machamer, C.E. The coronavirus e protein: Assembly and beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef] [Green Version]
- Ning, W.; Jiang, P.; Guo, Y.; Wang, C.; Tan, X.; Zhang, W.; Peng, D.; Xue, Y. Gps-palm: A deep learning-based graphic presentation system for the prediction of s-palmitoylation sites in proteins. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Wen, L.; Gao, X.; Jin, C.; Xue, Y.; Yao, X. Css-palm 2.0: An updated software for palmitoylation sites prediction. Protein Eng. Des. Sel. Peds 2008, 21, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Rottier, P.J.M. The coronavirus membrane glycoprotein. In The Coronaviridae; Siddell, S.G., Ed.; Springer: Boston, MA, USA, 1995; pp. 115–139. [Google Scholar]
- Kuo, L.; Hurst-Hess, K.R.; Koetzner, C.A.; Masters, P.S. Analyses of coronavirus assembly interactions with interspecies membrane and nucleocapsid protein chimeras. J. Virol. 2016, 90, 4357–4368. [Google Scholar] [CrossRef] [Green Version]
- Neuman, B.W.; Buchmeier, M.J. Supramolecular architecture of the coronavirus particle. Adv. Virus Res. 2016, 96, 1–27. [Google Scholar]
- Liu, P.; Jiang, J.Z.; Wan, X.F.; Hua, Y.; Li, L.; Zhou, J.; Wang, X.; Hou, F.; Chen, J.; Zou, J.; et al. Are pangolins the intermediate host of the 2019 novel coronavirus (sars-cov-2)? PLoS Pathog. 2020, 16, e1008421. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subir, R.; Jagat, J.M.; Kalyan, K.G. Pros and cons for use of statins in people with coronavirus disease-19 (covid-19). Diabetes Metab. Syndr. 2020, 14, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Peng, L. Letter in response to the article: Pros and cons for use of statins in 59 people with coronavirus disease-19 (covid-19)(ray, s et al.). Diabetes Metab. Syndr. 2020, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Mascitelli, L.; Grant, W.B.; Goldstein, M.R. Cholesterol levels, statins, vitamin d, and associated risk of pneumonia. Eur. J. Clin. Pharmacol. 2012, 68, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, L.; Meroni, L.; Galazzi, M.; Cesari, M.; Caramma, I.; Marchetti, G.; Galli, M.; Antinori, S. Does fluvastatin favour hcv replication in vivo? A pilot study on hiv-hcv coinfected patients. J. Viral Hepat. 2009, 16, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Verpaalen, B.; Neyts, J.; Delang, L. Are statins a viable option for the treatment of infections with the hepatitis c virus? Antivir. Res. 2014, 105, 92–99. [Google Scholar] [CrossRef]
- Blanchet, M.; Le, Q.T.; Seidah, N.G.; Labonté, P. Statins can exert dual, concentration dependent effects on hcv entry in vitro. Antivir. Res. 2016, 128, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S. Treating influenza infection, from now and into the future. Front Immunol 2018, 9, 1946. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of erk/mapk and pi3k/akt/mtor signaling modulation for middle east respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Appelberg, S.; Gupta, S.; Svensson Akusjärvi, S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Végvári, Á.; Benfeitas, R.; Sperk, M.; Ståhlberg, M.; et al. Dysregulation in akt/mtor/hif-1 signaling identified by proteo-transcriptomics of sars-cov-2 infected cells. Emerg. Microbes Infect. 2020, 9, 1748–1760. [Google Scholar] [CrossRef]
- Jones, S.F.; Infante, J.R. Molecular pathways: Fatty acid synthase. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Gan, T.; Zhang, J.; Ma, Q.; Liang, Y.; Zhao, Y. The structures and bioactivities of fatty acid synthase inhibitors. Curr. Med. Chem. 2019, 26, 7081–7101. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. Ampk: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. Srebp transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Bashmakov, Y.; Horton, J.D. Increased levels of nuclear srebp-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 1999, 274, 30028–30032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabe, D.; Xia, Z.P.; Adams, C.M.; Rawson, R.B. Three mutations in sterol-sensing domain of scap block interaction with insig and render srebp cleavage insensitive to sterols. Proc. Natl. Acad. Sci. USA 2002, 99, 16672–16677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Lee, J.N.; Lee, P.C.; Goldstein, J.L.; Brown, M.S.; Ye, J. Sterol-regulated ubiquitination and degradation of insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006, 3, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellaturu, C.R.; Deng, X.; Park, E.A.; Raghow, R.; Elam, M.B. Insulin enhances the biogenesis of nuclear sterol regulatory element-binding protein (srebp)-1c by posttranscriptional down-regulation of insig-2a and its dissociation from srebp cleavage-activating protein (scap).Srebp-1c complex. J. Biol. Chem. 2009, 284, 31726–31734. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. An emerging role of mtor in lipid biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. Mtor complex 1 regulates lipin 1 localization to control the srebp pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Majumdar, G.; O’Meally, R.N.; Cole, R.N.; Elam, M.B.; Raghow, R. Insulin-induced de novo lipid synthesis occurs mainly via mtor-dependent regulation of proteostasis of srebp-1c. Mol. Cell. Biochem. 2020, 463, 13–31. [Google Scholar] [CrossRef]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. Amp-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, E.A.; Tee, A.R. The kinase triad, ampk, mtorc1 and ulk1, maintains energy and nutrient homoeostasis. Biochem. Soc. Trans. 2013, 41, 939–943. [Google Scholar] [CrossRef]
- Han, Y.; Hu, Z.; Cui, A.; Liu, Z.; Ma, F.; Xue, Y.; Liu, Y.; Zhang, F.; Zhao, Z.; Yu, Y.; et al. Post-translational regulation of lipogenesis via ampk-dependent phosphorylation of insulin-induced gene. Nat. Commun. 2019, 10, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. Ampk phosphorylates and inhibits srebp activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorotea, D.; Koya, D.; Ha, H. Recent insights into srebp as a direct mediator of kidney fibrosis via lipid-independent pathways. Front. Pharmacol. 2020, 11, 265. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Hardie, D.G. Metabolism of inflammation limited by ampk and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.H.; Poffenberger, M.C.; Wong, A.H.; Jones, R.G. The role of ampk in t cell metabolism and function. Curr. Opin. Immunol. 2017, 46, 45–52. [Google Scholar] [CrossRef]
- Cameron, K.O.; Kurumbail, R.G. Recent progress in the identification of adenosine monophosphate-activated protein kinase (ampk) activators. Bioorganic Med. Chem. Lett. 2016, 26, 5139–5148. [Google Scholar] [CrossRef]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 aha/acc/tos guideline for the management of overweight and obesity in adults: A report of the american college of cardiology/american heart association task force on practice guidelines and the obesity society. J. Am. Coll. Cardiol. 2014, 63, 2985–3023. [Google Scholar] [CrossRef] [Green Version]
- Charakida, M.; Tousoulis, D.; Finer, N. Drug treatment of obesity in the cardiovascular patient. Curr. Opin. Cardiol. 2013, 28, 584–591. [Google Scholar] [CrossRef]
- Siebenhofer, A.; Jeitler, K.; Horvath, K.; Berghold, A.; Siering, U.; Semlitsch, T. Long-term effects of weight-reducing drugs in hypertensive patients. Cochrane Database Syst. Rev. 2013, Cd007654. [Google Scholar] [CrossRef]
- Yanovski, S.Z.; Yanovski, J.A. Long-term drug treatment for obesity: A systematic and clinical review. JAMA 2014, 311, 74–86. [Google Scholar] [CrossRef]
- Browne, C.D.; Hindmarsh, E.J.; Smith, J.W. Inhibition of endothelial cell proliferation and angiogenesis by orlistat, a fatty acid synthase inhibitor. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Pemble, C.W.t.; Johnson, L.C.; Kridel, S.J.; Lowther, W.T. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by orlistat. Nat. Struct. Mol. Biol. 2007, 14, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Guerciolini, R. Mode of action of orlistat. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1997, 21 (Suppl. 3), S12–S23. [Google Scholar]
- Zhi, J.; Mulligan, T.E.; Hauptman, J.B. Long-term systemic exposure of orlistat, a lipase inhibitor, and its metabolites in obese patients. J. Clin. Pharmacol. 1999, 39, 41–46. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin activates ampk through the lysosomal pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Hitakarun, A.; Khongwichit, S.; Wikan, N.; Roytrakul, S.; Yoksan, S.; Rajakam, S.; Davidson, A.D.; Smith, D.R. Evaluation of the antiviral activity of orlistat (tetrahydrolipstatin) against dengue virus, japanese encephalitis virus, zika virus and chikungunya virus. Sci. Rep. 2020, 10, 1499. [Google Scholar] [CrossRef] [Green Version]
- Bakhache, W.; Neyret, A.; McKellar, J.; Clop, C.; Bernard, E.; Weger-Lucarelli, J.; Briant, L. Fatty acid synthase and stearoyl-coa desaturase-1 are conserved druggable cofactors of old world alphavirus genome replication. Antivir. Res. 2019, 172, 104642. [Google Scholar] [CrossRef]
- Tongluan, N.; Ramphan, S.; Wintachai, P.; Jaresitthikunchai, J.; Khongwichit, S.; Wikan, N.; Rajakam, S.; Yoksan, S.; Wongsiriroj, N.; Roytrakul, S.; et al. Involvement of fatty acid synthase in dengue virus infection. Virol. J. 2017, 14, 28. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Singh, P.K.; Suhail, H.; Arumugaswami, V.; Pellett, P.E.; Giri, S.; Kumar, A. Amp-activated protein kinase restricts zika virus replication in endothelial cells by potentiating innate antiviral responses and inhibiting glycolysis. J. Immunol. 2020, 204, 1810–1824. [Google Scholar] [CrossRef]
- Esser, K.; Lucifora, J.; Wettengel, J.; Singethan, K.; Glinzer, A.; Zernecke, A.; Protzer, U. Lipase inhibitor orlistat prevents hepatitis b virus infection by targeting an early step in the virus life cycle. Antivir. Res. 2018, 151, 4–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Hood, B.L.; Chadwick, S.L.; Liu, S.; Watkins, S.C.; Luo, G.; Conrads, T.P.; Wang, T. Fatty acid synthase is up-regulated during hepatitis c virus infection and regulates hepatitis c virus entry and production. Hepatology 2008, 48, 1396–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xun, Y.H.; Zhang, Y.J.; Pan, Q.C.; Mao, R.C.; Qin, Y.L.; Liu, H.Y.; Zhang, Y.M.; Yu, Y.S.; Tang, Z.H.; Lu, M.J.; et al. Metformin inhibits hepatitis b virus protein production and replication in human hepatoma cells. J. Viral Hepat. 2014, 21, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Garcia-Valdecasas, M.; Gil-Gomez, A.; Rojas, A.; Gallego, P.; Ampuero, J.; Gallego-Duran, R.; Pastor, H.; Grande, L.; Padillo, F.J.; et al. Simvastatin and metformin inhibit cell growth in hepatitis c virus infected cells via mtor increasing pten and autophagy. PLoS ONE 2018, 13, e0191805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.L.; Chang, T.H.; Sun, W.C.; Chan, H.H.; Wu, C.C.; Hsu, P.I.; Cheng, J.S.; Yu, M.L. Metformin activates type i interferon signaling against hcv via activation of adenosine monophosphate-activated protein kinase. Oncotarget 2017, 8, 91928–91937. [Google Scholar] [CrossRef]
- Ammer, E.; Nietzsche, S.; Rien, C.; Kuhnl, A.; Mader, T.; Heller, R.; Sauerbrei, A.; Henke, A. The anti-obesity drug orlistat reveals anti-viral activity. Med Microbiol. Immunol. 2015, 204, 635–645. [Google Scholar] [CrossRef]
- Xie, W.; Wang, L.; Dai, Q.; Yu, H.; He, X.; Xiong, J.; Sheng, H.; Zhang, D.; Xin, R.; Qi, Y.; et al. Activation of ampk restricts coxsackievirus b3 replication by inhibiting lipid accumulation. J Mol Cell Cardiol 2015, 85, 155–167. [Google Scholar] [CrossRef]
- Samuelsson, L.; Gottsater, A.; Lindgarde, F. Decreasing levels of tumour necrosis factor alpha and interleukin 6 during lowering of body mass index with orlistat or placebo in obese subjects with cardiovascular risk factors. DiabetesObes. Metab. 2003, 5, 195–201. [Google Scholar] [CrossRef]
- Navina, S.; Acharya, C.; DeLany, J.P.; Orlichenko, L.S.; Baty, C.J.; Shiva, S.S.; Durgampudi, C.; Karlsson, J.M.; Lee, K.; Bae, K.T.; et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci. Transl. Med. 2011, 3, 107ra110. [Google Scholar] [CrossRef] [Green Version]
- Abou-El-Naga, I.F.; Said, D.E.; Gaafar, M.R.; Ahmed, S.M.; El-Deeb, S.A. A new scope for orlistat: Effect of approved anti-obesity drug against experimental microsporidiosis. Med Mycol. 2019, 57, 181–195. [Google Scholar] [CrossRef]
- Ursini, F.; Russo, E.; Pellino, G.; D’Angelo, S.; Chiaravalloti, A.; De Sarro, G.; Manfredini, R.; De Giorgio, R. Metformin and autoimmunity: A "new deal" of an old drug. Front Immunol 2018, 9, 1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunologic features in severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, J.; Isnard, S.; Lin, J.; Fombuena, B.; Marette, A.; Routy, B.; Chen, Y.; Routy, J.P. Metformin effect on gut microbiota: Insights for hiv-related inflammation. Aids Res. Ther. 2020, 17, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, M. The effects of metformin on gut microbiota and the immune system as research frontiers. Diabetologia 2017, 60, 1662–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor, V.M.; Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Lopez-Domenech, S.; Escribano-Lopez, I.; Rios-Navarro, C.; Alvarez, A.; Gomez, M.; Rocha, M.; et al. Metformin modulates human leukocyte/endothelial cell interactions and proinflammatory cytokines in polycystic ovary syndrome patients. Atherosclerosis 2015, 242, 167–173. [Google Scholar] [CrossRef]
- Li, C.Y.; Erickson, S.R.; Wu, C.H. Metformin use and asthma outcomes among patients with concurrent asthma and diabetes. Respirology 2016, 21, 1210–1218. [Google Scholar] [CrossRef] [Green Version]
- Calixto, M.C.; Lintomen, L.; Andre, D.M.; Leiria, L.O.; Ferreira, D.; Lellis-Santos, C.; Anhe, G.F.; Bordin, S.; Landgraf, R.G.; Antunes, E. Metformin attenuates the exacerbation of the allergic eosinophilic inflammation in high fat-diet-induced obesity in mice. PLoS ONE 2013, 8, e76786. [Google Scholar] [CrossRef]
- Park, C.S.; Bang, B.R.; Kwon, H.S.; Moon, K.A.; Kim, T.B.; Lee, K.Y.; Moon, H.B.; Cho, Y.S. Metformin reduces airway inflammation and remodeling via activation of amp-activated protein kinase. Biochem. Pharmacol. 2012, 84, 1660–1670. [Google Scholar] [CrossRef]
- Brandt, A.; Hernandez-Arriaga, A.; Kehm, R.; Sanchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 6668. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.J.; Kim, J.M.; Kim, I.K.; Ko, S.H.; Kim, J.S. Anti-inflammatory mechanism of metformin and its effects in intestinal inflammation and colitis-associated colon cancer. J. Gastroenterol. Hepatol. 2014, 29, 502–510. [Google Scholar] [CrossRef]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Lee, S.H.; Yang, E.J.; Kim, E.K.; Kim, J.K.; Shin, D.Y.; Cho, M.L. Metformin ameliorates inflammatory bowel disease by suppression of the stat3 signaling pathway and regulation of the between th17/treg balance. PLoS ONE 2015, 10, e0135858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, W.; Ding, Y.; Yu, X.; Ma, D.; Yang, B.; Li, Y.; Huang, L.; Chen, Z.; Zheng, J.; Yang, C. Metformin mitigates autoimmune insulitis by inhibiting th1 and th17 responses while promoting treg production. Am. J. Transl. Res. 2019, 11, 2393–2402. [Google Scholar] [PubMed]
- Filippatos, T.D.; Derdemezis, C.S.; Gazi, I.F.; Nakou, E.S.; Mikhailidis, D.P.; Elisaf, M.S. Orlistat-associated adverse effects and drug interactions: A critical review. Drug Saf. 2008, 31, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Lane, P.; Lee, K.; Parks, D. An integrated analysis of liver safety data from orlistat clinical trials. Obes. Facts 2012, 5, 485–494. [Google Scholar] [CrossRef]
- Powers, A.C. Diabetes mellitus: Diagnosis, classification, and pathophysiology. In Harrison’s Principles of Internal Medicine, 19th ed.; Kasper, D.L., Hauser, S.L., Jameson, J.L., Fauci, A.S., Longo, D.L., Loscalzo, J., Eds.; McGraw Hill: New York, NY, USA, 2015; Volume 2, p. 417e. [Google Scholar]
- Zhang, J.; Dong, J.; Martin, M.; He, M.; Gongol, B.; Marin, T.L.; Chen, L.; Shi, X.; Yin, Y.; Shang, F.; et al. Amp-activated protein kinase phosphorylation of angiotensin-converting enzyme 2 in endothelium mitigates pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 509–520. [Google Scholar] [CrossRef]
- Ursini, F.; Ciaffi, J.; Paola Landini, M.; Meliconi, R. Covid-19 and diabetes: Is metformin a friend or foe? Diabetes Res. Clin. Pract. 2020, 164, 108167. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ace2 activity impairs inactivation of des-arg9 bradykinin/bkb1r axis and facilitates lps-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ace2) in sars coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (adam17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (sars-cov) receptor, angiotensin-converting enzyme-2 (ace2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [Green Version]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of tnf-α-converting enzyme by the spike protein of sars-cov and ace2 induces tnf-α production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, W.; Grams, F.; Baumann, U.; Reinemer, P.; Gomis-Ruth, F.X.; McKay, D.B.; Bode, W. The metzincins--topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a superfamily of zinc-peptidases. Protein Sci. A Publ. Protein Soc. 1995, 4, 823–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, F.; Wang, Y.G.; Wang, C. Metformin inhibited growth, invasion and metastasis of esophageal squamous cell carcinoma in vitro and in vivo. Cell. Physiol. Biochem. 2018, 51, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; She, Z.G.; Cheng, X.; Qin, J.J.; Zhang, X.J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of blood glucose control and outcomes in patients with covid-19 and pre-existing type 2 diabetes. Cell Metab. 2020, 31, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Lalau, J.D.; Al-Salameh, A.; Hadjadj, S.; Goronflot, T.; Wiernsperger, N.; Pichelin, M.; Allix, I.; Amadou, C.; Bourron, O.; Duriez, T.; et al. Metformin use is associated with a reduced risk of mortality in patients with diabetes hospitalised for covid-19. Diabetes Metab. 2020, 101216. [Google Scholar] [CrossRef]
- Luo, P.; Qiu, L.; Liu, Y.; Liu, X.L.; Zheng, J.L.; Xue, H.Y.; Liu, W.H.; Liu, D.; Li, J. Metformin treatment was associated with decreased mortality in covid-19 patients with diabetes in a retrospective analysis. Am. J. Trop. Med. Hyg. 2020, 103, 69–72. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanner, J.E.; Alfieri, C. The Fatty Acid Lipid Metabolism Nexus in COVID-19. Viruses 2021, 13, 90. https://doi.org/10.3390/v13010090
Tanner JE, Alfieri C. The Fatty Acid Lipid Metabolism Nexus in COVID-19. Viruses. 2021; 13(1):90. https://doi.org/10.3390/v13010090
Chicago/Turabian StyleTanner, Jerome E., and Caroline Alfieri. 2021. "The Fatty Acid Lipid Metabolism Nexus in COVID-19" Viruses 13, no. 1: 90. https://doi.org/10.3390/v13010090
APA StyleTanner, J. E., & Alfieri, C. (2021). The Fatty Acid Lipid Metabolism Nexus in COVID-19. Viruses, 13(1), 90. https://doi.org/10.3390/v13010090