
Targeted Genome Sequencing (TG-Seq) Approaches to Detect Plant Viruses


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Extraction and cDNA Synthesis
2.3. Metagenomics Library Preparation (RNA-Seq)
2.4. Primer Design and PCR Optimization
2.5. Singleplex Amplicon PCR and Sequencing
2.6. Multiplex PCR (mPCR) and Sequencing (Include Negative Control Statement)
2.7. Serial Dilutions Multiplex PCR (mPCR) and Sequencing
2.8. Sequence Analysis
3. Results
3.1. RNA-Seq and TG-Seq Detection of Individual Monopartite, Bipartite and Tripartite Viruses
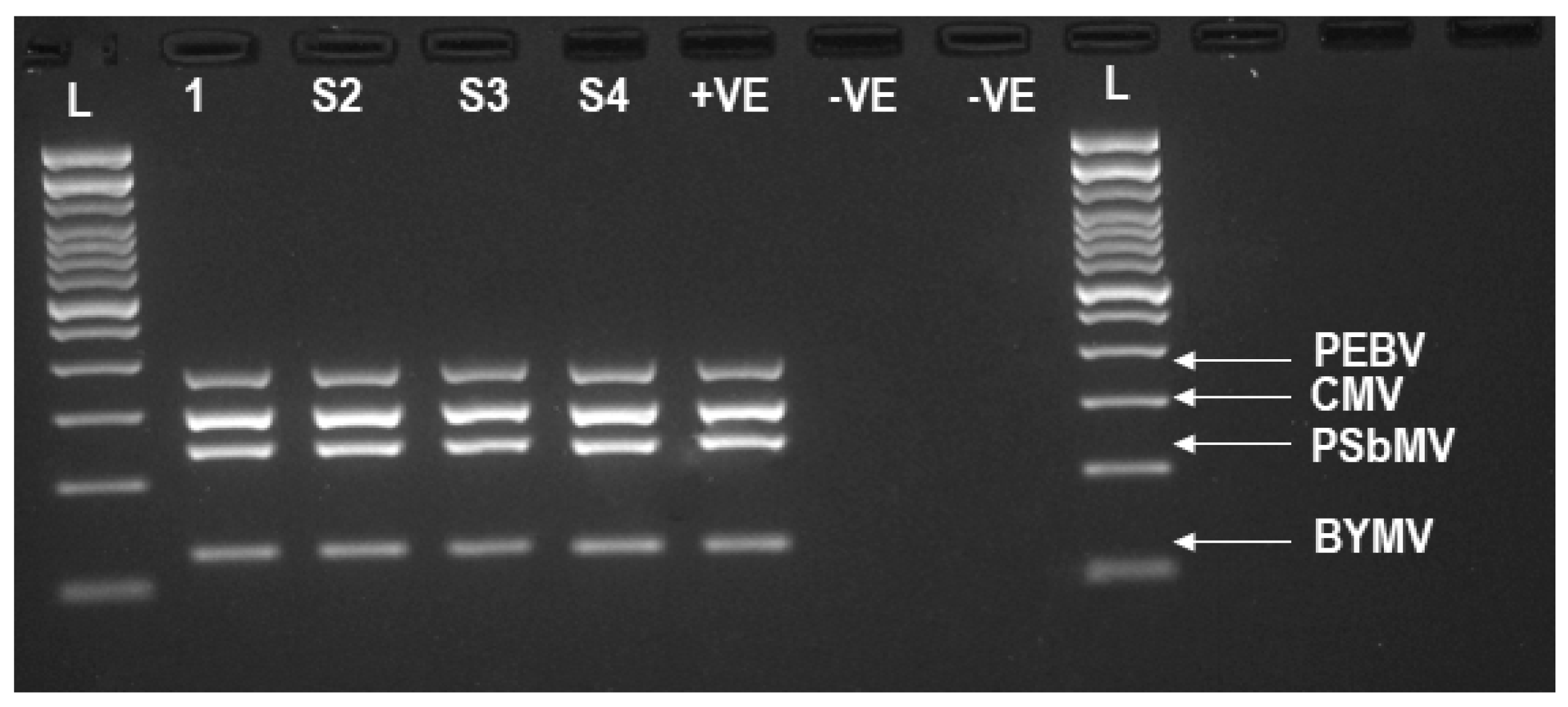
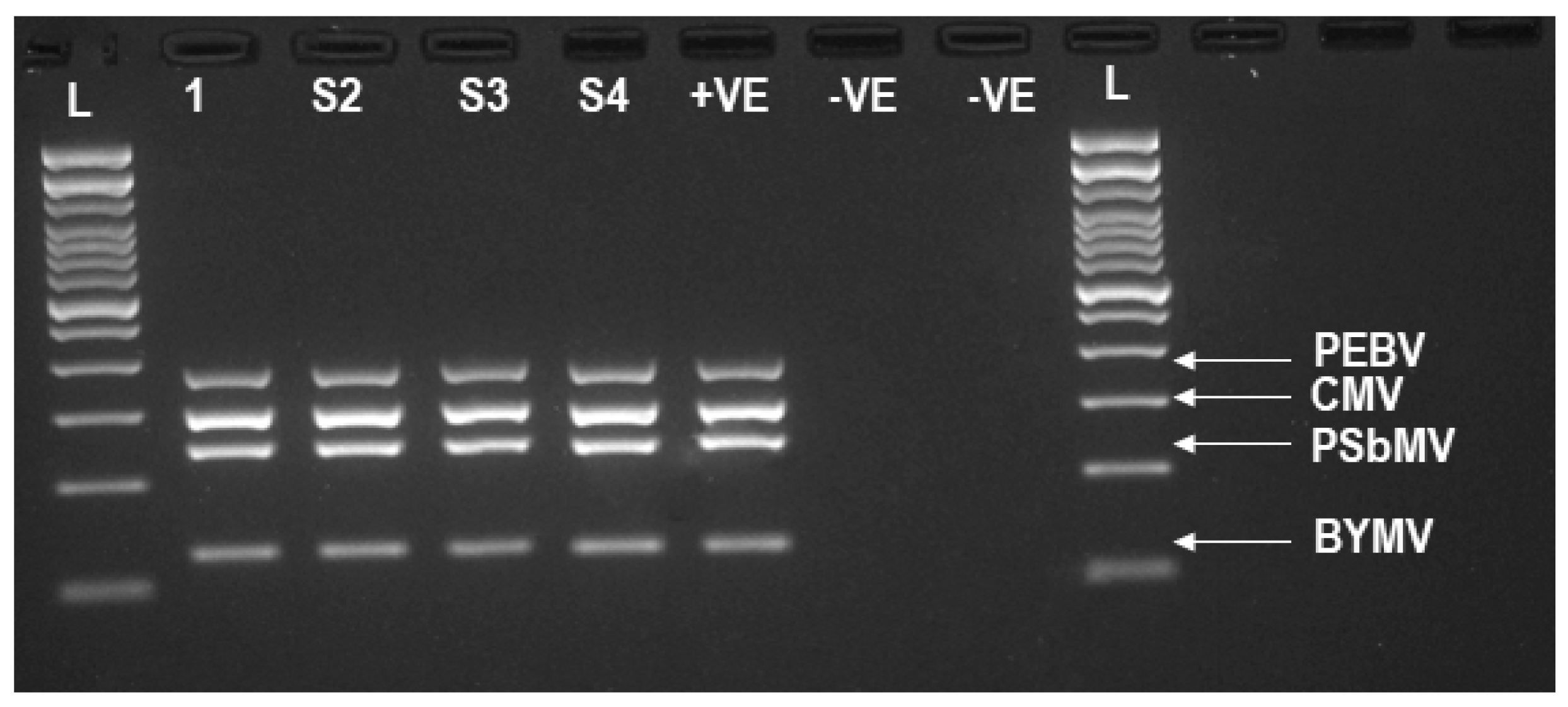
3.2. TG-Seq Detection of Multiple Monopartite, Bipartite and Tripartite Viruses in One Assay
3.3. Sensitivity of TG-Seq in Detecting Serially Diluted Multiple Viruses in One Assay
3.4. Sensitivity Comparison between RNA-Seq and TG-Seq as Detection Tools
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, R.A.C. Plant Virus Emergence and Evolution: Origins, New Encounter Scenarios, Factors Driving Emergence, Effects of Changing World Conditions, and Prospects for Control. Virus Res. 2009, 141, 113–130. [Google Scholar] [CrossRef]
- Jones, R.A.C.; Naidu, R.A. Global Dimensions of Plant Virus Diseases: Current Status and Future Perspectives. Annu. Rev. Virol. 2019, 6, 387–409. [Google Scholar] [CrossRef] [PubMed]
- Maina, S.; Barbetti, M.J.; Edwards, O.R.; de Almeida, L.; Ximenes, A.; Jones, R.A.C. Sweet Potato Feathery Mottle Virus and Sweet Potato Virus C from East Timorese and Australian Sweetpotato: Biological and Molecular Properties, and Biosecurity Implications. Am. Phytopath. Soc. 2018, 102, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Maina, S.; Coutts, B.A.; Edwards, O.R.; de Almeida, L.; Ximenes, A.; Jones, R.A.C. Papaya Ringspot Virus Populations From East Timorese and Northern Australian Cucurbit Crops: Biological and Molecular Properties, and Absence of Genetic Connectivity. Am. Phytopath. Soc. 2017, 101, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next Generation Sequencing for Detection and Discovery of Plant Viruses and Viroids: Comparison of Two Approaches. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete Viral Genome Sequence and Discovery of Novel Viruses by Deep Sequencing of Small RNAs: A Generic Method for Diagnosis, Discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Komínek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus detection by high-throughput sequencing of small RNAs: Large-scale performance testing of sequence analysis strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I.; Moscona, A. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. Am. Soc. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, L.; Galipienso, L.; Ferriol, I. Detection of Plant Viruses and Disease Management: Relevance of Genetic Diversity and Evolution. Front. Plant Sci. 2020. [Google Scholar] [CrossRef]
- Piper, A.M.; Batovska, J.; Cogan, N.O.I.; Weiss, J.; Cunningham, J.P.; Rodoni, B.C.; Blacket, M.J. Prospects and Challenges of Implementing DNA Metabarcoding for High-Throughput Insect Surveillance. Gigascience 2019, 8, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Mwaikono, K.S.; Maina, S.; Sebastian, A.; Schilling, M.; Kapur, V.; Gwakisa, P. High-Throughput Sequencing of 16S RRNA Gene Reveals Substantial Bacterial Diversity on the Municipal Dumpsite. BMC Microbiol. 2016, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Iqbal Wattoo, J.; Zafar Saleem, M.; Saqib Shahzad, M.; Arif, A.; Hameed, A.; Ahmad Saleem, M. DNA Barcoding: Amplification and Sequence Analysis of Rbcl and MatK Genome Regions in Three Divergent Plant Species. Adv. Life Sci. 2016, 4, 3–7. [Google Scholar]
- Pallás, V.; Sánchez-Navarro, J.A.; James, D. Recent Advances on the Multiplex Molecular Detection of Plant Viruses and Viroids. Front. Microb. 2018, 2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.S.; Bae, Y.J.; Jung, E.J.; Kang, S.J. RT-PCR-Based Detection of Six Garlic Viruses and Their Phylogenetic Relationships. J. Microbiol. Biotech. 2005, 15, 1110–1114. [Google Scholar]
- Zhao, X.; Liu, X.; Ge, B.; Li, M.; Hong, B. A Multiplex RT-PCR for Simultaneous Detection and Identification of Five Viruses and Two Viroids Infecting Chrysanthemum. Arch. Virol. 2015, 160, 1145–1152. [Google Scholar] [CrossRef]
- Kwak, H.R.; Kim, M.K.; Shin, J.C.; Lee, Y.J.; Seo, J.K.; Lee, H.U.; Jung, M.N.; Kim, S.H.; Choi, H.S. The Current Incidence of Viral Disease in Korean Sweet Potatoes and Development of Multiplex RT-PCR Assays for Simultaneous Detection of Eight Sweet. Plant Pathol. J. 2014, 30, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambino, G. Multiplex Rt-Pcr Method for the Simultaneous Detection of Nine Grapevine Viruses. Methods Mol. Biol. 2015, 1236, 39–47. [Google Scholar] [CrossRef]
- Sánchez-Navarro, J.A.; Aparicio, F.; Herranz, M.C.; Minafra, A.; Myrta, A.; Pallás, V. Simultaneous Detection and Identification of Eight Stone Fruit Viruses by One-Step RT-PCR. Eur. J. Plant Pathol. 2005, 111, 77–84. [Google Scholar] [CrossRef]
- Kinoti, W.M.; Constable, F.E.; Nancarrow, N.; Plummer, K.M.; Rodoni, B. Analysis of Intra-Host Genetic Diversity of Prunus Necrotic Ringspot Virus (PNRSV) Using Amplicon next Generation Sequencing. PLoS ONE 2017, 12, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Meekins, D.A.; Trujillo, J.D.; Gaudreault, N.N.; Morozov, I.; Pérez-Núñez, D.; Revilla, Y.; Richt, J.A. Long amplicon sequencing for improved genetic characterization of African swine fever virus. J. Virol. Methods 2020, 285, 113946. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and IVar. Genome Biol. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford-Siltz, L.A.; Mullis, L.; Sanad, Y.M.; Tohma, K.; Lepore, C.J.; Azevedo, M.; Parra, G.I. Genomics Analyses of GIV and GVI Noroviruses Reveal the Distinct Clustering of Human and Animal Viruses. Viruses 2019, 11, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F. Trim Galore. 2015. Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore (accessed on 19 March 2021). (accessed from Agriculture Victoria computational system (BASC).
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method withreduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The Metagenomics RAST Server—A Public Resource for the Automatic Phylogenetic and Functional Analysis of Metagenomes. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef] [Green Version]
- Maina, S.; Zheng, L.; King, S.; Mcqueen, V.L.; Norton, S.L.; Rodoni, B. Transcriptome Sequencing Reveals the Genome Sequence of Pea Early Browning Virus from a 29-Year-Old Faba Bean Sample. Am. Soc. Microbiol. 2020. [Google Scholar] [CrossRef]
- Maina, S.; Zheng, L.; King, S.; Aftab, M.; Nancarrow, N.; Trębicki, P.; Rodoni, B. Genome Sequence and Phylogeny of a Bean Yellow Mosaic Virus Isolate Obtained from a 14-Year-Old Australian Lentil Sample. Am. Soc. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Man, J.; Wu, Y. Development of a multiplex polymerase chain reaction for simultaneous detection of wheat viruses and a phytoplasma in China. Arch. Virol. 2012, 157, 1261–1267. [Google Scholar] [CrossRef]
- Kwon, J.Y.; Hong, J.S.; Kim, M.J.; Choi, S.H.; Min, B.E.; Song, E.G.; Kim, H.H.; Ryu, K.H. Simultaneous Multiplex PCR Detection of Seven Cucurbit-Infecting Viruses. J. Virol. Methods 2014, 206, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.; Lee, Y.H.; Park, C.Y.; Lee, M.A.; Bae, Y.S.; Lim, S.; Lee, J.H.; Moon, J.S.; Lee, S.H. Development of Multiplex RT-PCR for Simultaneous Detection of Garlic Viruses and the Incidence of Garlic Viral Disease in Garlic Genetic Resources. Plant Pathol. J. 2015, 31, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Cheng, J.; Huang, T.; Zheng, X.; Wu, Y. A multiplex reverse transcription PCR assay for simultaneous detection of five tobacco viruses in tobacco plants. J. Virol. Methods 2012, 183, 57–62. [Google Scholar] [CrossRef]
- Liu, H.; Wu, K.; Wu, W.; Mi, W.; Hao, X.; Wu, Y. A Multiplex Reverse Transcription PCR Assay for Simultaneous Detection of Six Main RNA Viruses in Tomato Plants. J. Virol. Methods 2019, 265, 53–58. [Google Scholar] [CrossRef]
- Tuo, D.; Shen, W.; Yang, Y.; Yan, P.; Li, X.; Zhou, P. Development and Validation of a Multiplex Reverse Transcription PCR Assay for Simultaneous Detection of Three Papaya Viruses. Viruses 2014, 6, 3893–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, P.; Shen, W.T.; Yan, P.; Tuo, D.C.; Li, X.Y.; Zhou, P. Simultaneous Detection of Papaya Ringspot Virus, Papaya Leaf Distortion Mosaic Virus, and Papaya Mosaic Virus by Multiplex Real-Time Reverse Transcription PCR. Acta Virol. 2015, 59, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Ellis, M.D.; Hoak, J.M.; Ellis, B.W.; Brown, J.A.; Sit, T.L.; Wilkinson, C.A.; David Reed, T.; Welbaum, G.E. Quantitative Real-Time PCR Analysis of Individual Flue-Cured Tobacco Seeds and Seedlings Reveals Seed Transmission of Tobacco Mosaic Virus. Am. Phytopath. Soc. 2020, 110, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Anitha, S.; Monyo, E.S.; Okori, P. Simultaneous Detection of Groundnut Rosette Assistor Virus (GRAV), Groundnut Rosette Virus (GRV) and Satellite RNA (SatRNA) in Groundnuts Using Multiplex RT-PCR. Arch. Virol. 2014, 159, 3059–3062. [Google Scholar] [CrossRef]
- Maurastoni, M.; Sá-Antunes, T.F.; Oliveira, S.A.; Santos, A.M.; Ventura, J.A.; Fernandes, P.M. A multiplex RT-PCR method to detect papaya meleira virus complex in adult pre-flowering plants. Arch. Virol. 2020, 165, 1211–1214. [Google Scholar] [CrossRef]
- Boonham, N.; Tomlinson, J.; Mumford, R. Microarrays for Rapid Identification of Plant Viruses. Annu. Rev. Phytopathol. 2007, 45, 307–328. [Google Scholar] [CrossRef]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High Throughput Sequencing for Plant Virus Detection and Discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.H.; Braxton, C.; Eloit, M.; Feng, S.F.; Fragnoud, R.; Mallet, L.; Mee, E.T.; Sathiamoorthy, S.; Vandeputte, O.; Khan, A.S. Current Perspectives on High-Throughput Sequencing (HTS) for Adventitious Virus Detection: Upstream Sample Processing and Library Preparation. Viruses 2018, 10, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into Biases and Sequencing Errors for Amplicon Sequencing with the Illumina MiSeq Platform. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Takahashi, C.N.; Organick, L.; Bee, C.; Ang, S.D.; Weiss, P.; Peck, B.; Seelig, G.; Ceze, L.; Strauss, K. Quantifying Molecular Bias in DNA Data Storage. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.S.; Emerson, R.O.; Sherwood, A.M.; Desmarais, C.; Chung, M.W.; Parsons, J.M.; Steen, M.S.; LaMadrid-Herrmannsfeldt, M.A.; Williamson, D.W.; Livingston, R.J.; et al. Using Synthetic Templates to Design an Unbiased Multiplex PCR Assay. Nat. Commun. 2013, 4, 2680. [Google Scholar] [CrossRef] [Green Version]
- Kinoti, W.M.; Constable, F.E.; Nancarrow, N.; Plummer, K.M.; Rodoni, B. Generic Amplicon Deep Sequencing to Determine Ilarvirus Species Diversity in Australian Prunus. Front. Microbiol. 2017, 8, 1219. [Google Scholar] [CrossRef]
- Bruinsma, S.; Burgess, J.; Schlingman, D.; Czyz, A.; Morrell, N.; Ballenger, C.; Meinholz, H.; Brady, L.; Khanna, A.; Freeberg, L.; et al. Bead-Linked Transposomes Enable a Normalization-Free Workflow for NGS Library Preparation. BMC Genom. 2018, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology 2015, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Ding, S.W.; Zhang, Y.; Zhu, S. Identification of Viruses and Viroids by Next-Generation Sequencing and Homology-Dependent and Homology-Independent Algorithms. Ann. Rev. Phytopathol. 2015, 425–444. [Google Scholar] [CrossRef]
- Adams, I.; Fox, A. Diagnosis of Plant Viruses Using Next-Generation Sequencing and Metagenomic Analysis. Curr. Res. Top. Plant Virol. 2016, 323–335. [Google Scholar] [CrossRef]
- Morlan, J.D.; Qu, K.; Sinicropi, D.V. Selective Depletion of rRNA Enables Whole Transcriptome Profiling of Archival Fixed Tissue. PLoS ONE 2012, 7, e42882. [Google Scholar] [CrossRef]
- Nagano, A.J.; Honjo, M.N.; Mihara, M.; Sato, M.; Kudoh, H. Detection of Plant Viruses in Natural Environments by Using RNA-Seq. Methods Mol. Biol. 2015, 1236, 89–98. [Google Scholar] [CrossRef]
- Gaafar, Y.Z.A.; Ziebell, H. Comparative Study on Three Viral Enrichment Approaches Based on RNA Extraction for Plant Virus/Viroid Detection Using High-Throughput Sequencing. PLoS ONE 2020, 15, e0237951. [Google Scholar] [CrossRef]
- Ndunguru, J.; Sseruwagi, P.; Tairo, F.; Stomeo, F.; Maina, S.; Djinkeng, A.; Kehoe, M.; Boykin, L.M.; Melcher, U. Analyses of Twelve New Whole Genome Sequences of Cassava Brown Streak Viruses and Ugandan Cassava Brown Streak Viruses from East Africa: Diversity, Supercomputing and Evidence for Further Speciation. PLoS ONE 2015, 10, e0139321. [Google Scholar] [CrossRef] [Green Version]
- Guerret, M.G.L.; Nyalugwe, E.P.; Maina, S.; Barbetti, M.J.; van Leur, J.A.G.; Jones, R.A.C. Biological and Molecular Properties of a Turnip Mosaic Virus (TuMV) Strain That Breaks TuMV Resistances in Brassica Napus. Plant. Dis. 2017, 101, 674–683. [Google Scholar] [CrossRef]
- Havlová, K.; Fajkus, J. G4 structures in control of replication and transcription of rRNA genes. Front. Plant Sci. 2020, 11, 593692. [Google Scholar] [CrossRef]
- MacConaill, L.E.; Burns, R.T.; Nag, A.; Coleman, H.A.; Slevin, M.K.; Giorda, K.; Light, M.; Lai, K.; Jarosz, M.; McNeill, M.S.; et al. Unique, Dual-Indexed Sequencing Adapters with UMIs Effectively Eliminate Index Cross-Talk and Significantly Improve Sensitivity of Massively Parallel Sequencing. BMC Genom. 2018, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Costello, M.; Fleharty, M.; Abreu, J.; Farjoun, Y.; Ferriera, S.; Holmes, L.; Granger, B.; Green, L.; Howd, T.; Mason, T.; et al. Characterization and Remediation of Sample Index Swaps by Non-Redundant Dual Indexing on Massively Parallel Sequencing Platforms. BMC Genom. 2018, 19, 332. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.E.; Boone, B.E. Next-Generation Sequencing Strategies. Cold Spring Harb. Perspect. Med. 2019, 9, a025791. [Google Scholar] [CrossRef] [PubMed]
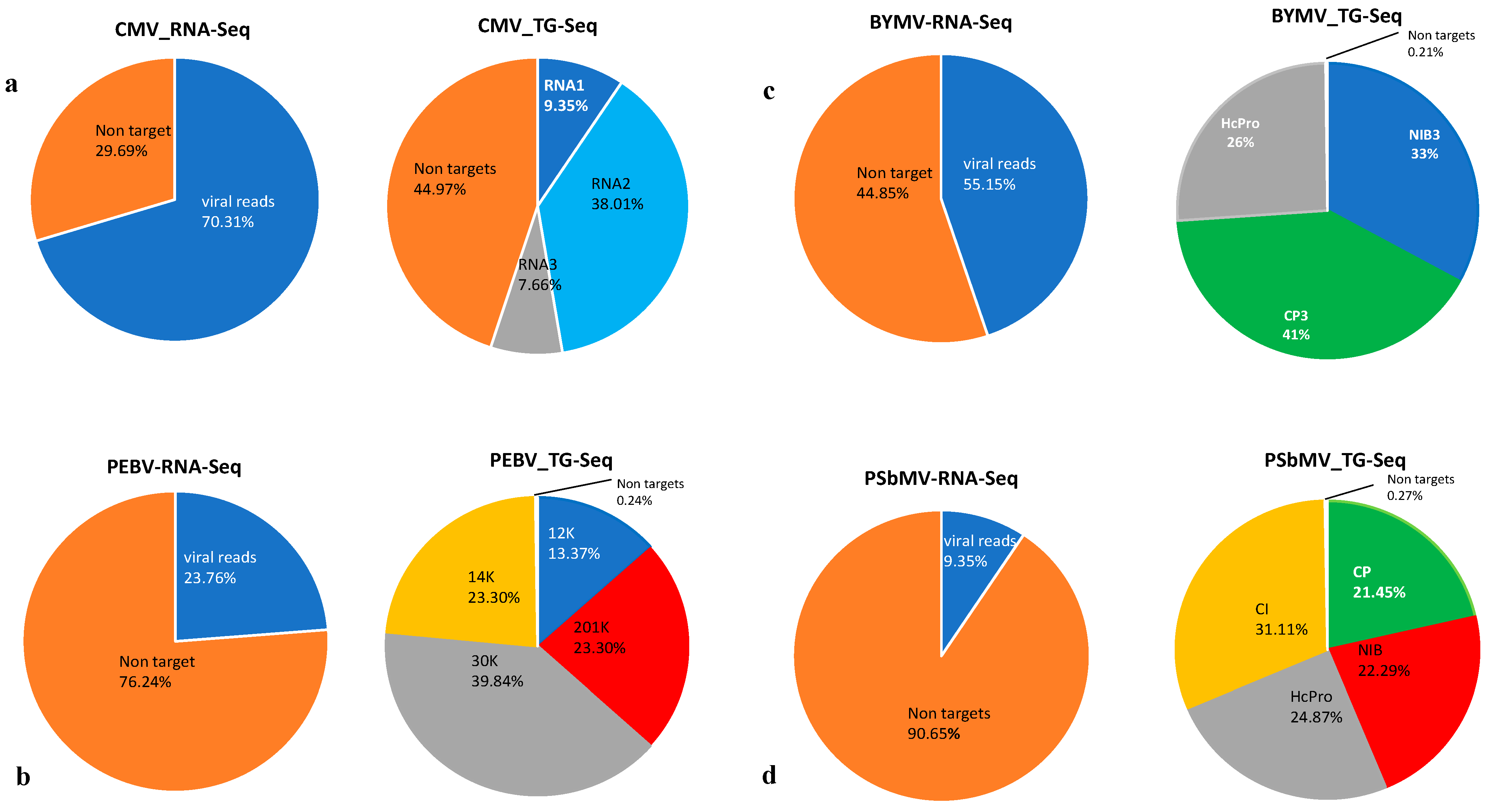


{kind=link}
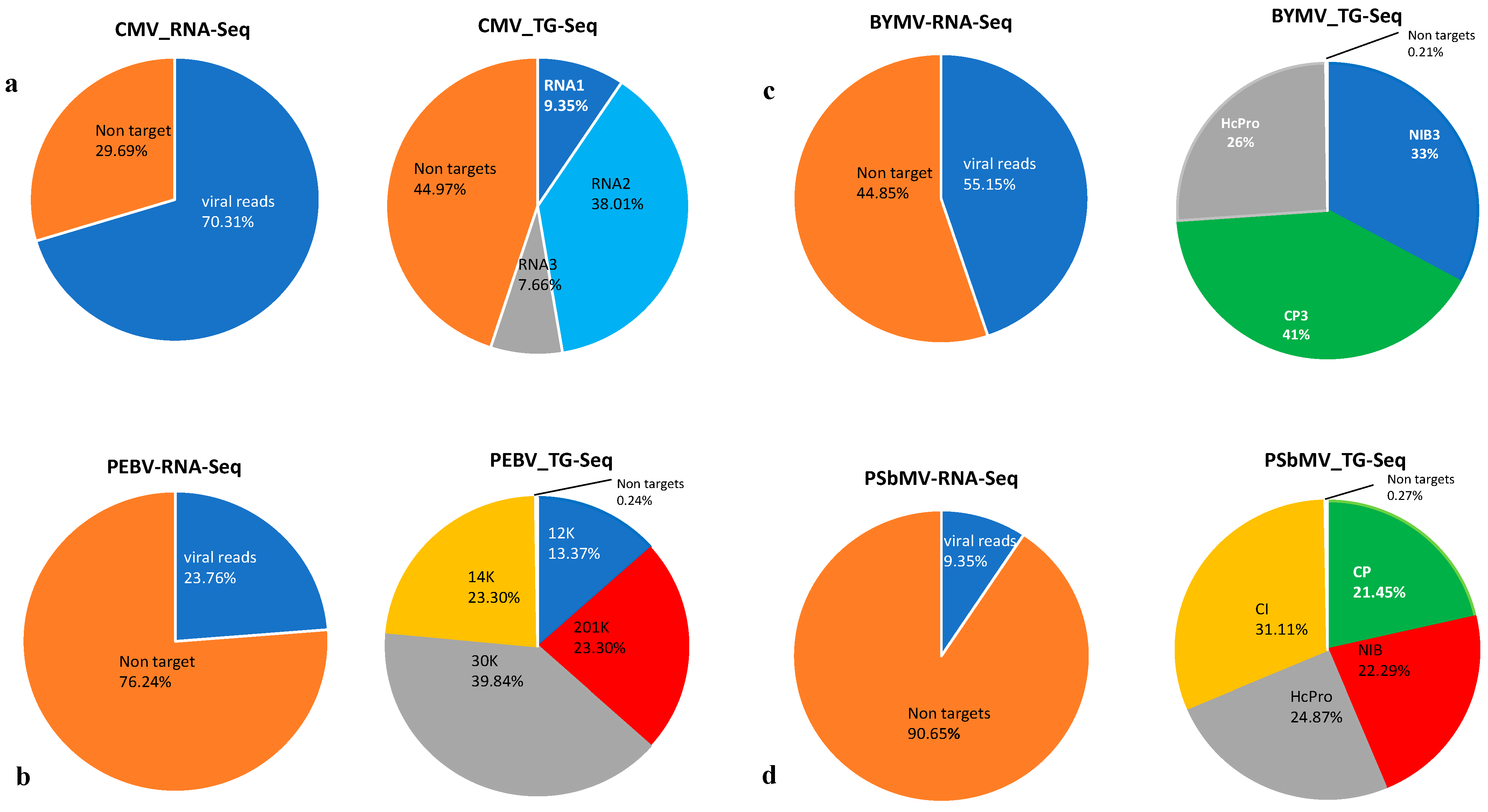{kind=link}
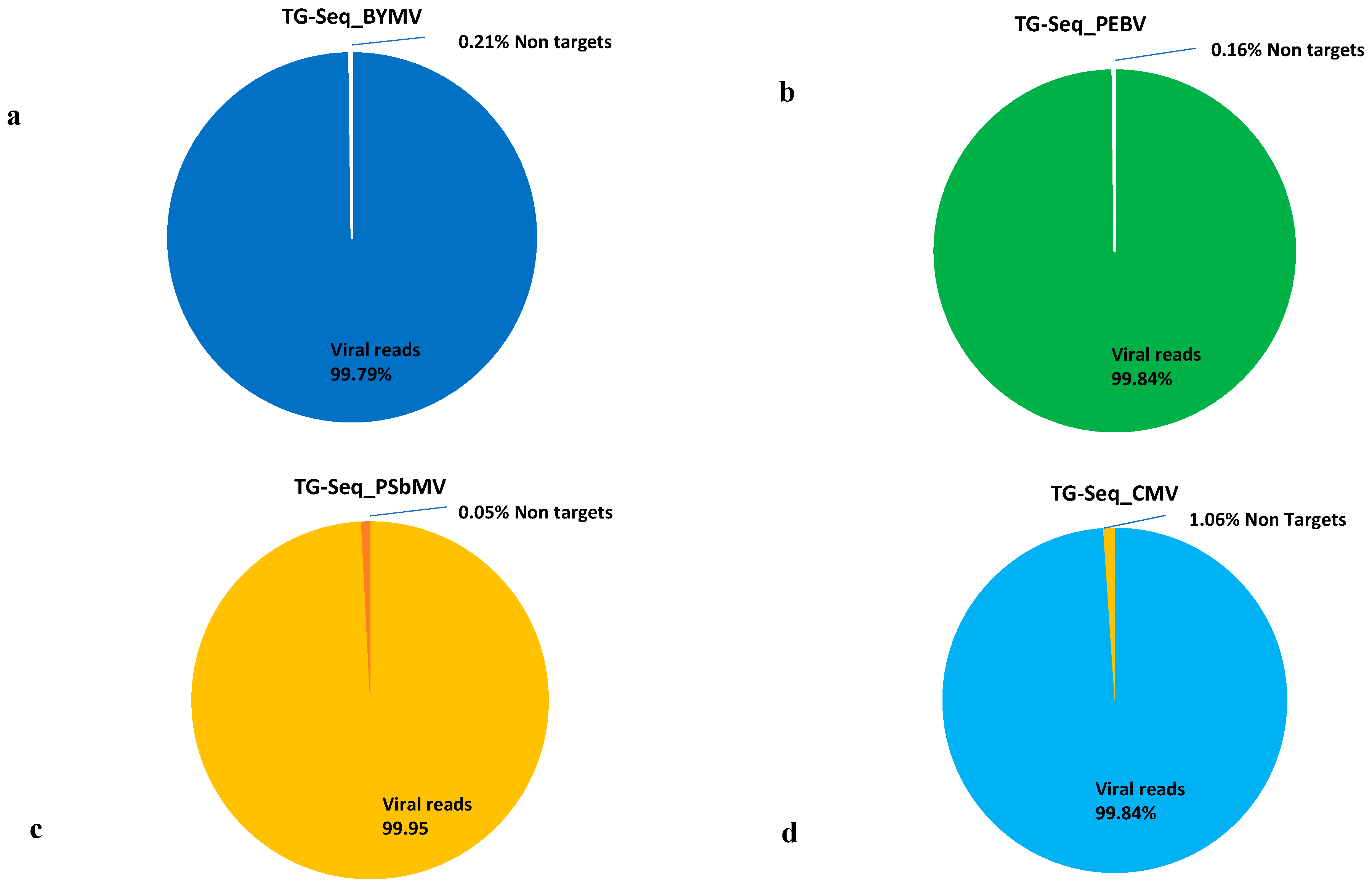{kind=link}
| Sample | Host | Virus | Coverage (x) c | No of Read Counts Mapping to the Virus | GC Content | Genome Size | GenBank Accession |
|---|---|---|---|---|---|---|---|
| 14BY a | Lentil | BYMV | 2307 | 1,331,893 | 39.4% | 9868 | LC500882 |
| 13C | Field pea | PSbMV | 718 | 507,189 | 41.5% | 9852 | SRR13206509 |
| LY-2 b | Faba bean | PEBV-RNA1 | 3899 | 235,443 | 40.7% | 7037 | LC528622 |
| LY-2 b | Faba bean | PEBV-RNA2 | 5606 | 1,252,888 | 42% | 2604 | LC528623 |
| 14C | Faba bean | CMV-RNA1 | 8774 | 318,290 | 45.3% | 3215 | SRR13197436 |
| 14C | Faba bean | CMV-RNA2 | 37,144 | 1,293,807 | 45.3% | 2892 | SRR13197436 |
| 14C | Faba bean | CMV-RNA3 | 10,615 | 260,700 | 47.1% | 2188 | SRR13197436 |
| Primer | Target Virus | Target Genome Region | Amplicon | Primer Sequence (5′-3′) | Amplicon Size (bp) | Optimal Annealing Temperature (Tm; (°C) | Primer Position Binding Site |
|---|---|---|---|---|---|---|---|
| HcPro-1F a | BYMV | HcPro | HcProF1 | CCTTGTGGTCGTATCACTTGTAA | 132 | 64.4 | 1182–1204 |
| HcPro-1R a | CTGAATGGTGCCTCTGGTAAC | 64.9 | 1412–1432 | ||||
| BYHcProF2 | BYMV | HcPro | HcProF2 | CCTTGTGGTCGTATCACTTGTAA | 251 | 64.4 | 1199–1222 |
| BYHcProR2 | CTGAATGGTGCCTCTGGTAAC | 64.9 | 1429–1449 | ||||
| BYNIb2F | BYMV | NIb | NIb2 | AGAGCAATTCAACCAGAGCATAG | 283 | 64.9 | 8247–8269 |
| BYNIb2R | CACAAGCACCTCATCAGTCTC | 64.9 | 8505–8525 | ||||
| BYNIb3F | BYMV | NIb | NIb3 | TTACAGCCGCACCGATTG | 288 | 64.9 | 7549–7566 |
| BYNIb3R | CGCATCTCAAGAACAGCATTC | 65 | 7766–7786 | ||||
| BYCPF3 | BYMV | CP | CPF3 | GAATGGACAATGATGGATGGAGAG | 287 | 65.2 | 8966–8989 |
| BYCPR3 | CTAACTGCTGCCGCCTTC | 65 | 9235–9252 | ||||
| HCPF2 | PSbMV | HcPro | HcPro | AGTTAGGCATCTGGCAATAG | 359 | 61.3 | 2028–2047 |
| HCPR2 | AGTCCTTAGCATCCTTCTCA | 61.8 | 2367–2386 | ||||
| CI-1F | PSbMV | CI | CI | TTGCGTGATTCGTCTATGC | 296 | 62.4 | 5227–5245 |
| CI-1R | TGTGCTATCGTTCTTGTAATTGA | 62.3 | 5500–5522 | ||||
| NIbF3 | PSbMV | NIb | NIb | GTGCGTCCAGATTGTGAA | 328 | 61.8 | 8338–8355 |
| NIbR3 | TACTTCTATATGGCTCCTGTTCTA | 62 | 8642–8665 | ||||
| PCP-F1 a | PSbMV | CP | CP | GAACATCAGGAACCATCACA | 254 | 61.7 | 9005–9024 |
| PCP-R1 a | TTCAATACACCACACCATCAA | 60.4 | 9238–9259 | ||||
| 12K2F | PEBV | 12K | 12K | GAAGTGTGCTGTGTCAAC | 294 | 60.4 | 6279–6296 |
| 12K2R | AAACCGAAATCTATGTCATCTC | 60.1 | 6551–6572 | ||||
| 14KF4 | PEBV | 14K | 14K | AGATGTGGACGACTCAGTGAA | 254 | 65 | 2303–2323 |
| 14KR4 | CGAAGTTGGCGAAGTGGTT | 65.1 | 2538–2556 | ||||
| 30KF | PEBV | 30K | 30K | TCATCGTAGAAGAGAGACTGTGTT | 348 | 65 | 5626–5649 |
| 30KR | ACCGCAACCGTACCTATCT | 64.7 | 5955–5973 | ||||
| 201K-F a | PEBV | 201K | 201K | GGTTAGAAGTGCTGGAAGTGAA | 399 | 64.4 | 1621–1642 |
| 201K-R a | TCATTGGCTTGCGACTCTC | 64.3 | 2001–2019 | ||||
| CMVRNA1F a | CMV | RNA1 | RNA1 | CTCCCACGGCGATAAAGG | 315 | 57.56 | 133–150 |
| CMVRNA1R a | GTGACCCAACTTCCTCCGA | 58.94 | 429–447 | ||||
| CMVRNA2F | CMV | RNA2 | RNA2 | ATAACMTCCCAGTTCTCACC | 260 | 56.23 | 1488–1507 |
| CMVRNA2R | TGRAARTCRCACCACCAYTT | 57.25 | 1728–1747 | ||||
| CMVRNA3F | CMV | RNA3 | RNA3 | GAAATTYGATTCRACYGTGTGGG | 202 | 58.02 | 1601–1623 |
| CMVRNA3R | CTTNCKCATRTCRCCDATATCAGC | 56.98 | 1779–1802 |
| Library | Amplicons Targeted by mPCR a | Raw Reads | No. of Reads after QC (%) | Amplicons Detected by TG-Seq b | Amplicons of BYMV, PSbMV, PEBV and CMV, Detected by GE |
|---|---|---|---|---|---|
| 1 | BYMV (NIb2, CPF3, HcProF2) | 3,754,078 | 97.74% | NIb (1,208,788), CP (1,503,144), HcPro (949,413) | CP, HcPro |
| 2 | BYMV (HcProF2, NIb3, CPF3) | 3,704,546 | 98.07% | HcPro (1,748,342), NIb (22,057), CP (1,839,520) | CP, HcPro |
| 3 | BYMV (NIb2, CP3, HcProF1) | 3,563,538 | 98.04% | NIb (1,487,879), CP (1,091,068), HcPro (906,692) | HcPro, NIb, CP |
| 4 | BYMV (CP, HcProF1, HcProF2) | 3,523,172 | 97.85% | CP (880,456), HcPro (2,490,101) | CP, HcPro |
| 5 | PSbMV (CP, NIb, HcPro, CI) | 4,568,980 | 98.71% | CP (967,475), NIb (1,005,493), HcPro (1,121,760), CI (1,403,227) | CP, NIb, HcPro, CI |
| 6 | PEBV (12K, 14K, 30K, 201K) | 4,110,734 | 98.46% | 12K (706,420), 14K (979,796), 30K (1,371,956), 201K (978,813) | 12K, 14K, 30K, 201K |
| 7 | PEBV (12K, 14K, 30K, 201K) | 3,923,838 | 98.50% | 12K (515,527), 14K (900,724), 30K (153,998), 201K (899,718) | 12K, 14K, 30K, 201K |
| 8 | CMV (RNA1, RNA2, RNA3) | 3,457,376 | 98.44% | RNA1 (318,290), RNA2 (1,293,807), RNA3 (260,700) | RNA1, RNA2 |
| 9 | CMV (RNA1), PEBV (201K), PSbMV (CP) | 3,257,938 | 98.21% | RNA1 (1,299,800), 201K (1,145,237), CP (732,277) | RNA1, 201K, CP |
| 10 | CMV(RNA3), PEBV (201K2), PSbMV (HcPro), BYMV (CP3) | 3,318,404 | 98.37% | RNA3 (207), 201K (1,561,718), HcPro (419,226, CP (1,248,550) | 201K, HcPro, CP3 |
| 11 | CMV (RNA1), PEBV (201K), PSbMV (CP), BYMV (HcPro) | 2,210,396 | 95.24% | RNA1 (703,928), 201K (8929), CP (5348), HcPro (1,057,739) | RNA1, HcPro |
| 12 | CMV (RNA1), PEBV (201K), PSbMV (CP), BYMV (HcPro) | 2,514,042 | 98.54% | RNA1 (735,687), 201K (701,502), CP (645,571), HcPro (739,571) | RNA1, 201K, CP, HcPro |
| Library | Virus | mPCR Product Concentration | Raw Reads | No. of Reads after QC (%) | Virus Amplicons Detected by TG-Seq | Amplicons Detected by GE |
|---|---|---|---|---|---|---|
| 10−2 | CMV,PEBV,PSbMV,BYMV | 16.9 ng/uL | 2,245,566 | 98.05% | RNA1 (1,465,542), 201K (130,757), CP (2,224), HcPro (519,838) | RNA1, 201K, HcPro |
| 10−4 | CMV,PEBV,PSbMV,BYMV | 8 ng/uL | 2,332,290 | 97.49% | RNA1 (1,831,035), 201K (91,395), CP (27,503), HcPro (239,682) | RNA1, HcPro |
| 10−6 | CMV,PEBV,PSbMV,BYMV | 8 ng/uL | 1,924,302 | 96.71% | RNA1 (807,712), 201K (74,276), CP (246,891), HcPro (724,792) | RNA1 *, HcPro * |
| 10−8 | CMV,PEBV,PSbMV,BYMV | 7 ng/uL | 2,221,416 | 96.45% | RNA1 (1,096,272), (201K) 127,154, CP (210, 534), HcPro (704,258) | RNA1 *, HcPro * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maina, S.; Zheng, L.; Rodoni, B.C. Targeted Genome Sequencing (TG-Seq) Approaches to Detect Plant Viruses. Viruses 2021, 13, 583. https://doi.org/10.3390/v13040583
Maina S, Zheng L, Rodoni BC. Targeted Genome Sequencing (TG-Seq) Approaches to Detect Plant Viruses. Viruses. 2021; 13(4):583. https://doi.org/10.3390/v13040583
Chicago/Turabian StyleMaina, Solomon, Linda Zheng, and Brendan C. Rodoni. 2021. "Targeted Genome Sequencing (TG-Seq) Approaches to Detect Plant Viruses" Viruses 13, no. 4: 583. https://doi.org/10.3390/v13040583
APA StyleMaina, S., Zheng, L., & Rodoni, B. C. (2021). Targeted Genome Sequencing (TG-Seq) Approaches to Detect Plant Viruses. Viruses, 13(4), 583. https://doi.org/10.3390/v13040583