
Impaired Antiviral Responses to Extracellular Double-Stranded RNA and Cytosolic DNA, but Not to Interferon-α Stimulation, in TRIM56-Deficient Cells


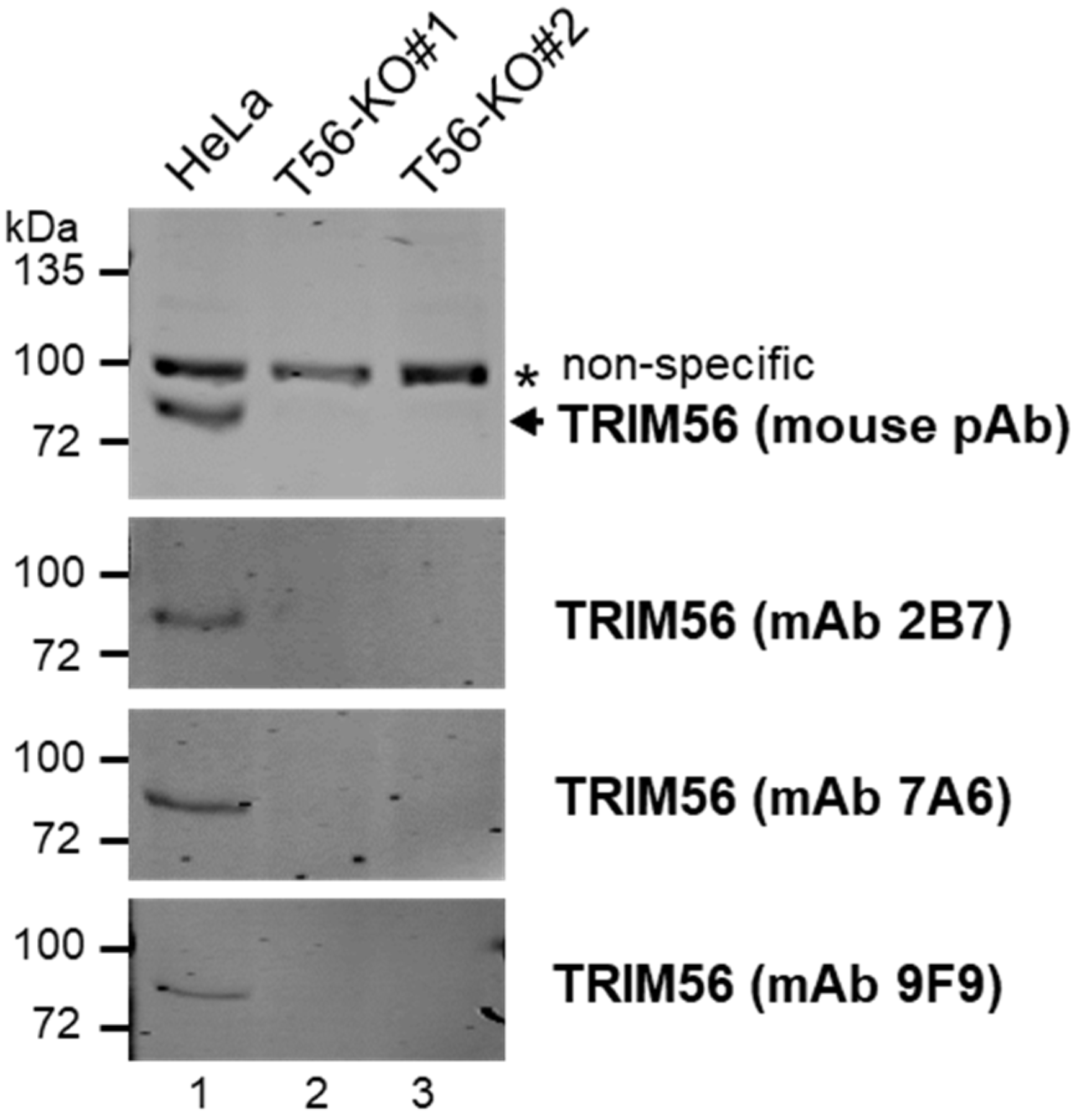{kind=link}
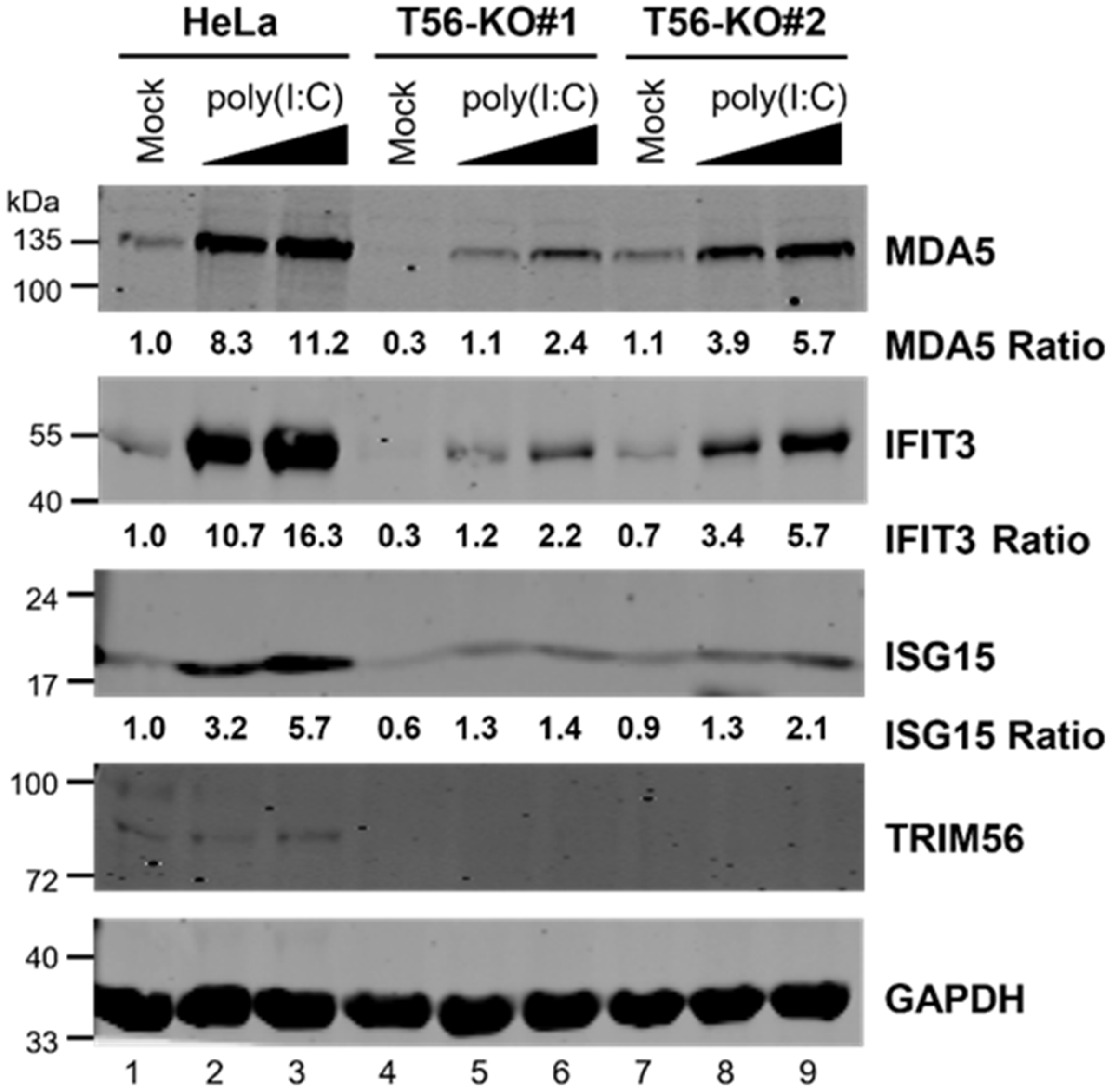{kind=link}
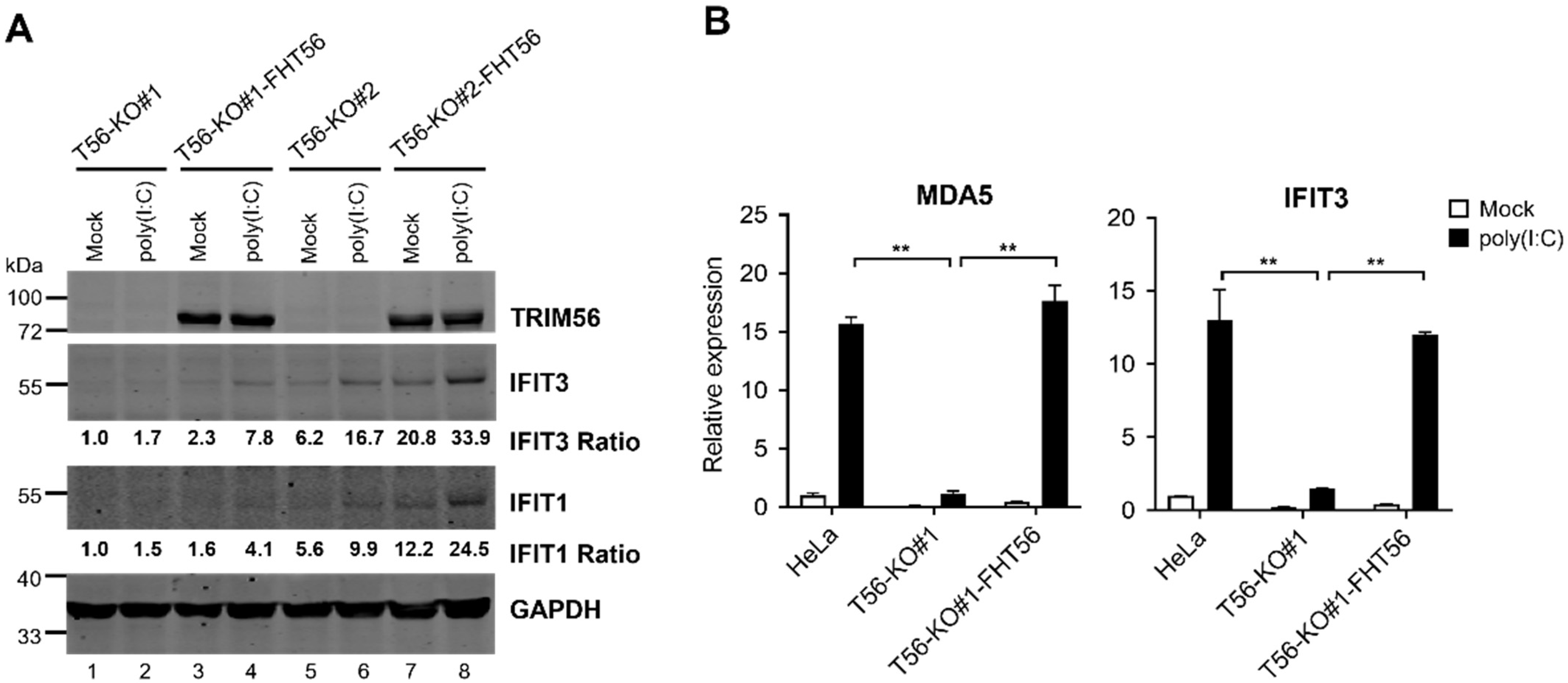{kind=link}
{kind=link}
{kind=link}
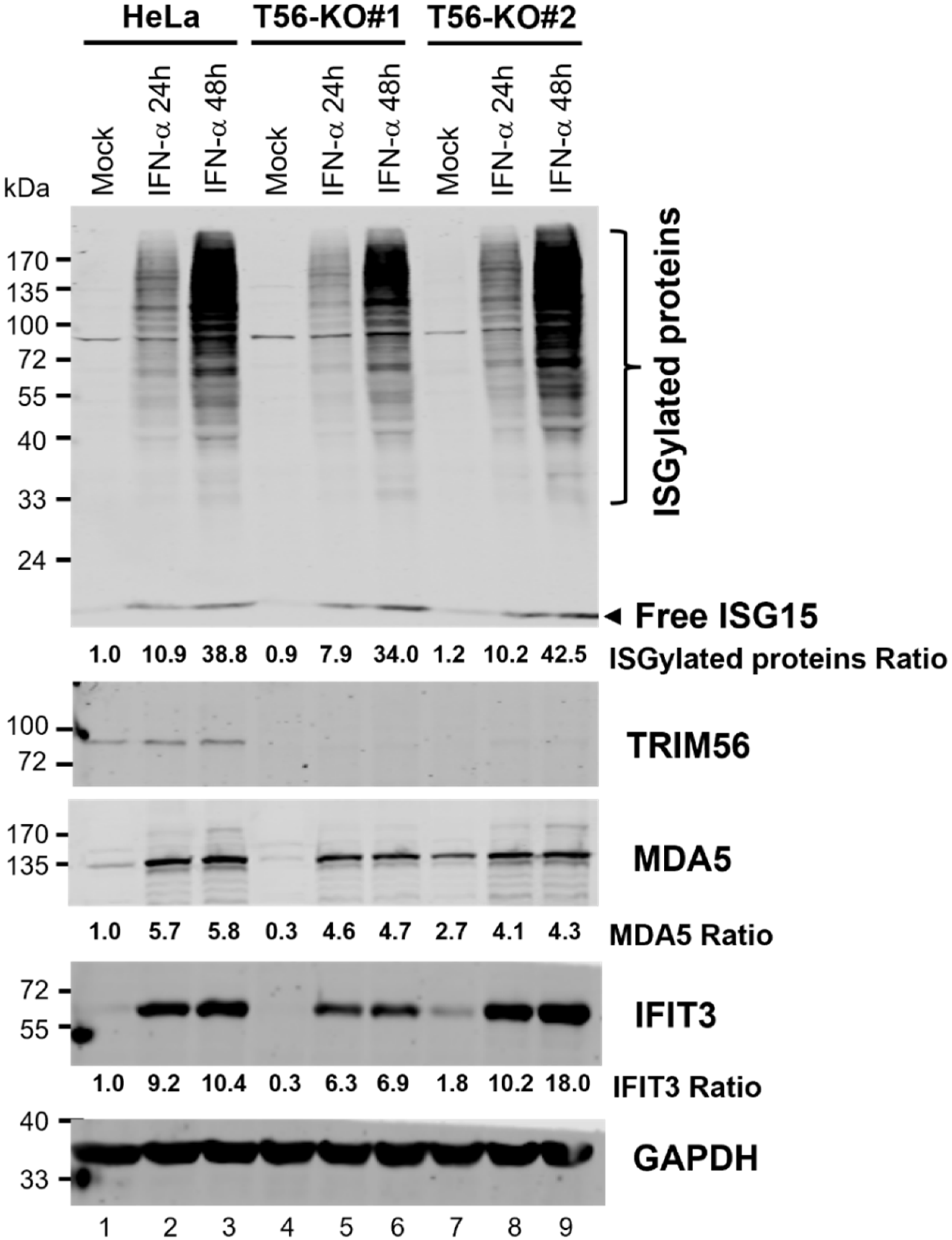{kind=link}
{kind=link}
{kind=link}
Abstract
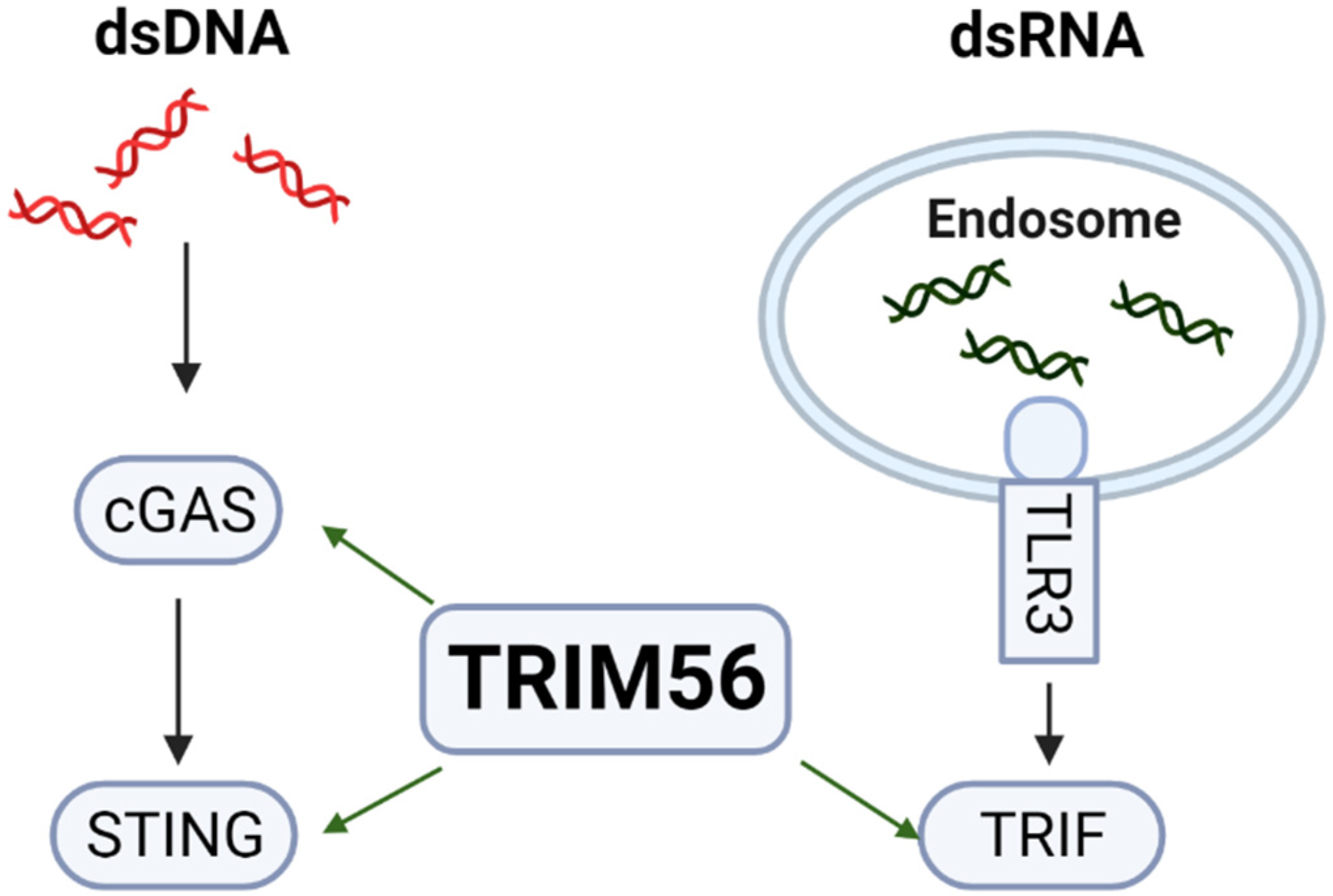
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Lines
2.3. Pattern Recognition Receptor Ligands, IFN-α, and VSV-Luc
2.4. Stimulation of Cells and Antiviral Activity Assay
2.5. Quantitative PCR
2.6. Protein Analyses
2.7. Statistical Analysis
3. Results
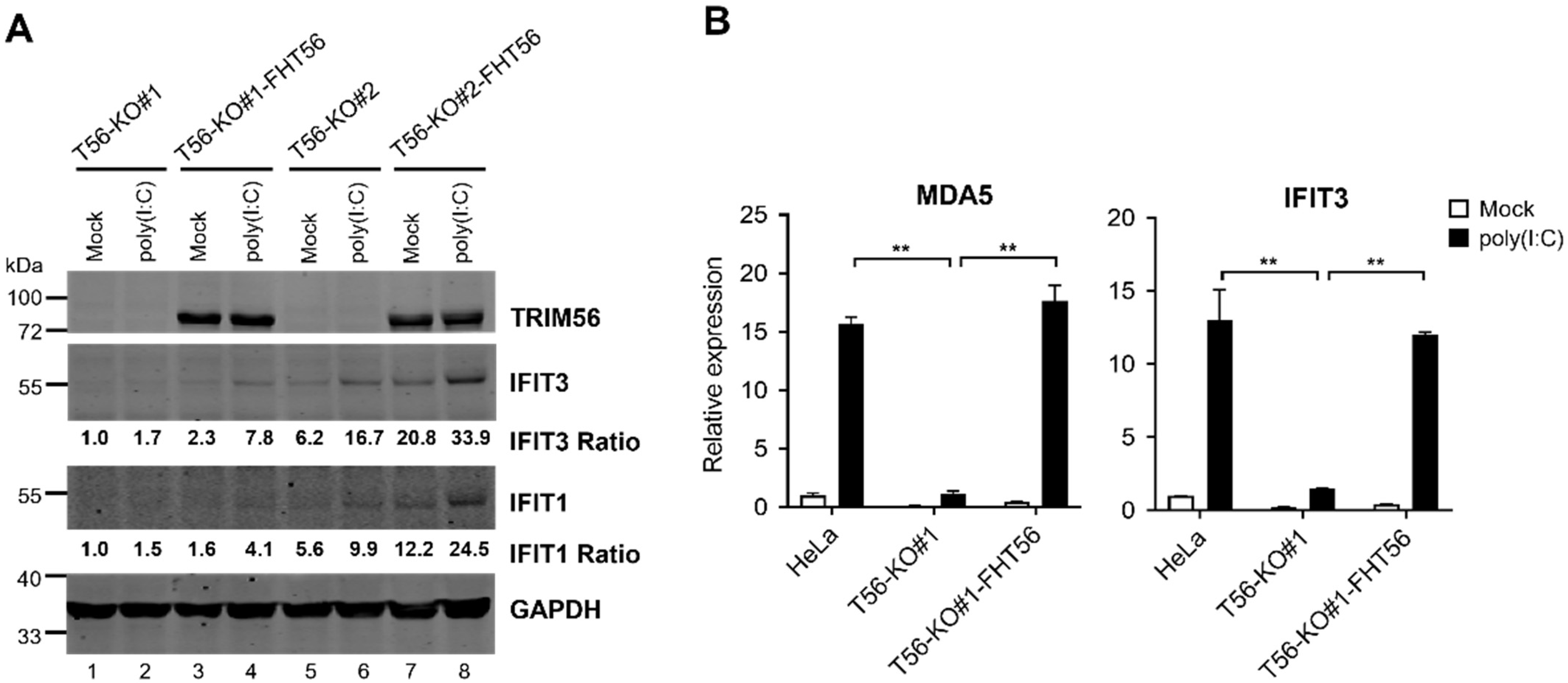
3.1. Knockout of TRIM56 Severely Compromises, but Does Not Eliminate Extracellular dsRNA-Induced Antiviral Gene Expression
3.2. Reconstitution of TRIM56 Expression in HeLa T56-KO Cell Lines Reverses the Impaired TLR3 Response Phenotype
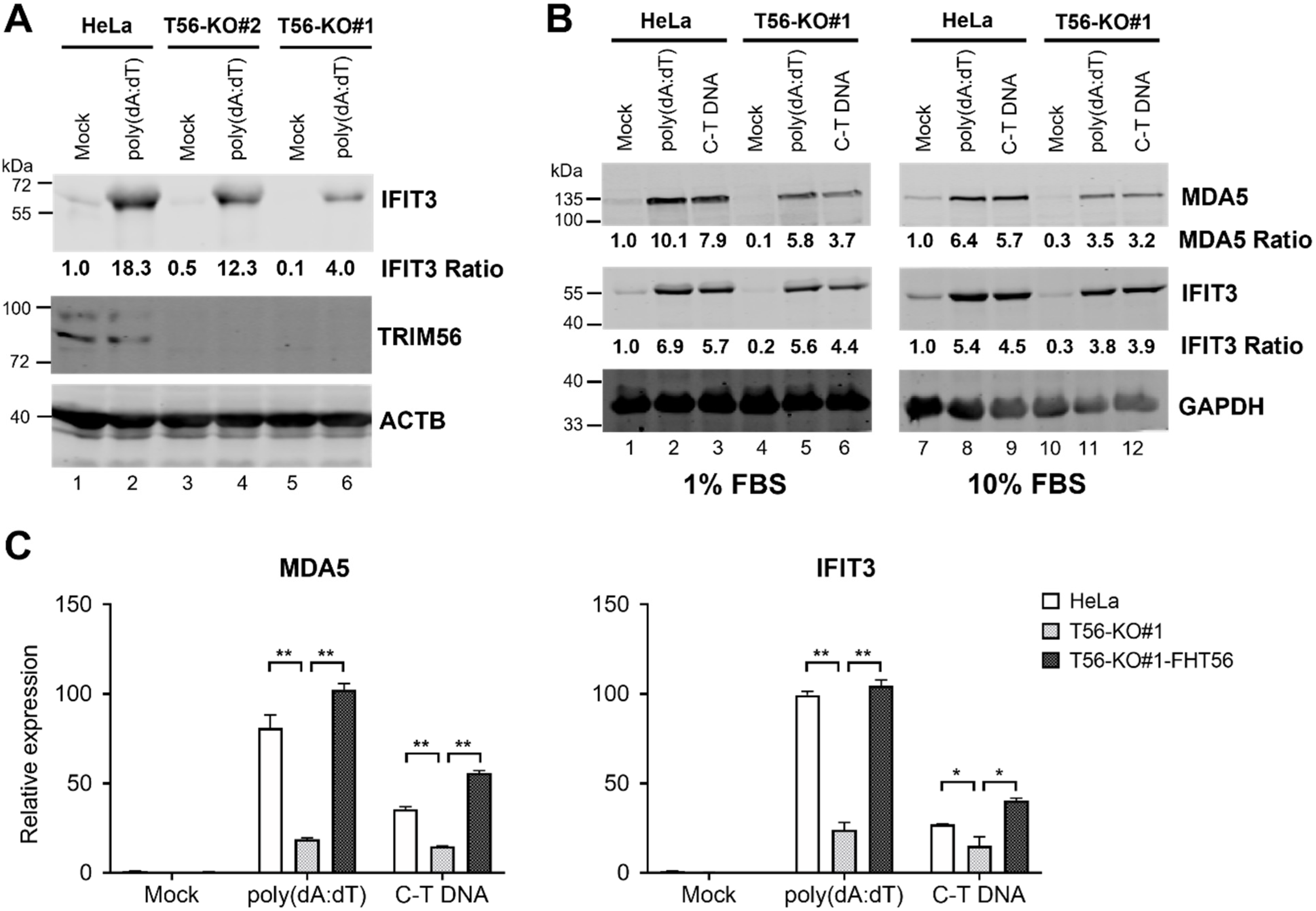
3.3. TRIM56 Deficiency Is Associated with Reduced ISG Response to Cytosolic dsDNA
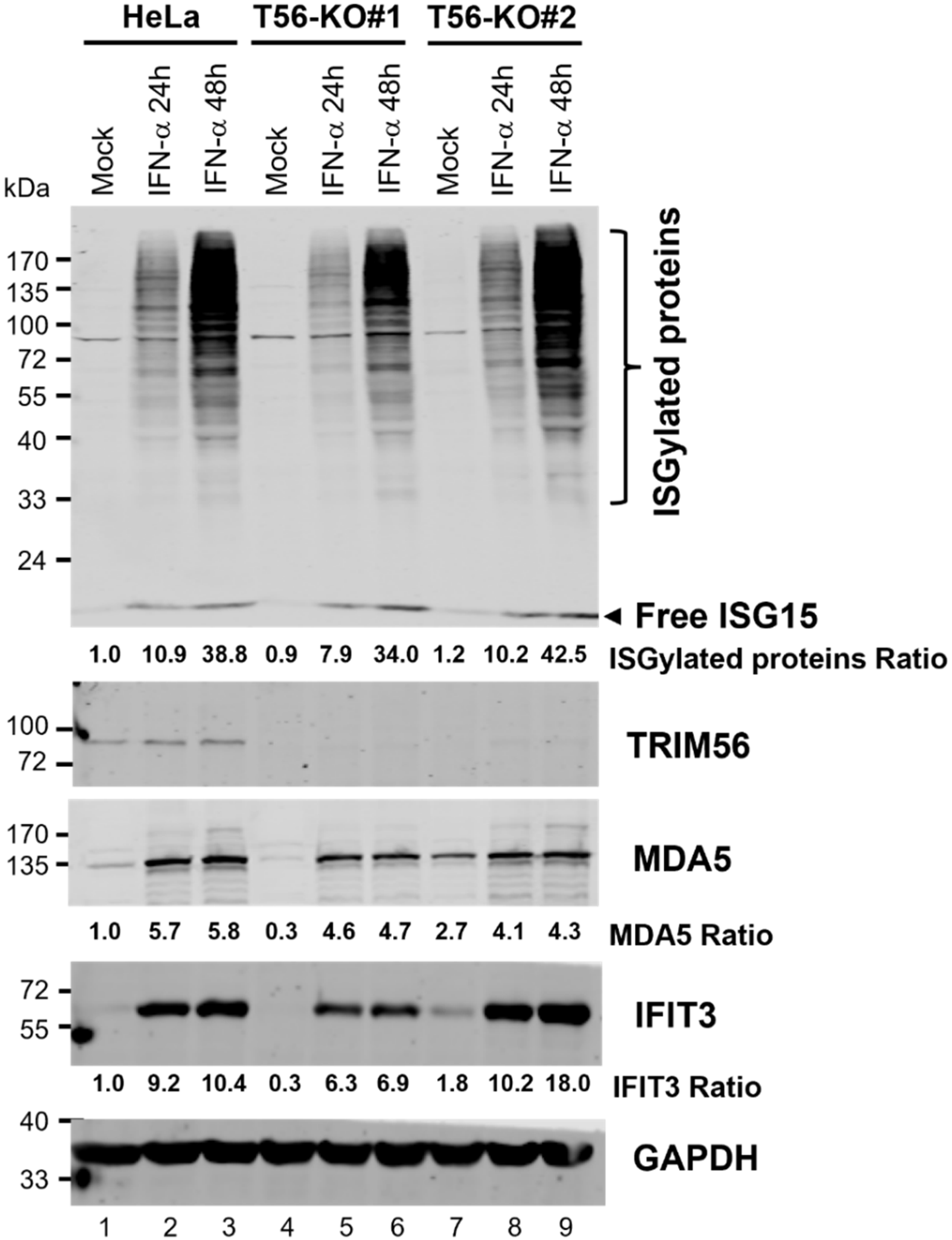
3.4. TRIM56 Deletion Does Not Impair ISG Induction by IFN-α
3.5. ISGylation Takes Place Efficiently in the Absence of TRIM56
3.6. TRIM56 Is Not Required for the Establishment of an Antiviral State by IFN-α
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Munir, M. TRIM proteins: Another class of viral victims. Sci. Signal. 2010, 3, jc2. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, B.; Wang, N.; Lee, Y.M.; Liu, C.; Li, K. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J. Virol. 2011, 85, 3733–3745. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 2010, 33, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Li, N.L.; Wang, J.; Liu, B.; Lester, S.; Li, K. TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J. Biol. Chem. 2012, 287, 36404–36413. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.J.; Kim, C.; Shin, W.J.; Sklan, E.H.; Eoh, H.; Jung, J.U. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 2018, 9, 613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajsbaum, R.; Garcia-Sastre, A.; Versteeg, G.A. TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 2014, 426, 1265–1284. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, N.L.; Wang, J.; Shi, P.Y.; Wang, T.; Miller, M.A.; Li, K. Overlapping and distinct molecular determinants dictating the antiviral activities of TRIM56 against flaviviruses and coronavirus. J. Virol. 2014, 88, 13821–13835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Li, N.L.; Wei, D.; Liu, B.; Guo, F.; Elbahesh, H.; Zhang, Y.; Zhou, Z.; Chen, G.Y.; Li, K. The E3 ligase TRIM56 is a host restriction factor of Zika virus and depends on its RNA-binding activity but not miRNA regulation, for antiviral function. PLoS Negl. Trop. Dis. 2019, 13, e0007537. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, N.L.; Shen, Y.; Bao, X.; Fabrizio, T.; Elbahesh, H.; Webby, R.J.; Li, K. The C-Terminal Tail of TRIM56 Dictates Antiviral Restriction of Influenza A and B Viruses by Impeding Viral RNA Synthesis. J. Virol. 2016, 90, 4369–4382. [Google Scholar] [CrossRef] [Green Version]
- Kane, M.; Zang, T.M.; Rihn, S.J.; Zhang, F.; Kueck, T.; Alim, M.; Schoggins, J.; Rice, C.M.; Wilson, S.J.; Bieniasz, P.D. Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell Host. Microbe 2016, 20, 392–405. [Google Scholar] [CrossRef] [Green Version]
- Cureton, D.K.; Massol, R.H.; Saffarian, S.; Kirchhausen, T.L.; Whelan, S.P. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 2009, 5, e1000394. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Dong, Q.; Li, J.; Jangra, R.K.; Fan, M.; Brasier, A.R.; Lemon, S.M.; Pfeffer, L.M.; Li, K. Viral induction of the zinc finger antiviral protein is IRF3-dependent but NF-kappaB-independent. J. Biol. Chem. 2010, 285, 6080–6090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumthip, K.; Yang, D.; Li, N.L.; Zhang, Y.; Fan, M.; Sethuraman, A.; Li, K. Pivotal role for the ESCRT-II complex subunit EAP30/SNF8 in IRF3-dependent innate antiviral defense. PLoS Pathog. 2017, 13, e1006713. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Swaminathan, S. Human IFIT proteins inhibit lytic replication of KSHV: A new feed-forward loop in the innate immune system. PLoS Pathog. 2019, 15, e1007609. [Google Scholar] [CrossRef]
- Wang, N.; Liang, Y.; Devaraj, S.; Wang, J.; Lemon, S.M.; Li, K. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J. Virol. 2009, 83, 9824–9834. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, T.; Du, M.; Chen, X.; Du, F.; Ren, J.; Chen, Z.J. Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 2021, 371, eabc5386. [Google Scholar] [CrossRef]
- Li, K.; Chen, Z.; Kato, N.; Gale, M., Jr.; Lemon, S.M. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 2005, 280, 16739–16747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, R.; Wang, C.; Jiang, Q.; Lv, M.; Gao, P.; Yu, X.; Mu, P.; Zhang, R.; Bi, S.; Feng, J.M.; et al. NEMO-IKKbeta Are Essential for IRF3 and NF-kappaB Activation in the cGAS-STING Pathway. J. Immunol. 2017, 199, 3222–3233. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, D.E. Interferon-stimulated gene 15 and the protein ISGylation system. J. Interferon Cytokine Res. 2011, 31, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Zhang, D.E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Liu, T.T.; Lin, H.; Zhang, M.; Wei, J.; Luo, W.W.; Hu, Y.H.; Zhong, B.; Hu, M.M.; Shu, H.B. TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLoS Pathog. 2017, 13, e1006600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Wang, R.; Li, K. Impaired Antiviral Responses to Extracellular Double-Stranded RNA and Cytosolic DNA, but Not to Interferon-α Stimulation, in TRIM56-Deficient Cells. Viruses 2022, 14, 89. https://doi.org/10.3390/v14010089
Wang D, Wang R, Li K. Impaired Antiviral Responses to Extracellular Double-Stranded RNA and Cytosolic DNA, but Not to Interferon-α Stimulation, in TRIM56-Deficient Cells. Viruses. 2022; 14(1):89. https://doi.org/10.3390/v14010089
Chicago/Turabian StyleWang, Dang, Ruixue Wang, and Kui Li. 2022. "Impaired Antiviral Responses to Extracellular Double-Stranded RNA and Cytosolic DNA, but Not to Interferon-α Stimulation, in TRIM56-Deficient Cells" Viruses 14, no. 1: 89. https://doi.org/10.3390/v14010089
APA StyleWang, D., Wang, R., & Li, K. (2022). Impaired Antiviral Responses to Extracellular Double-Stranded RNA and Cytosolic DNA, but Not to Interferon-α Stimulation, in TRIM56-Deficient Cells. Viruses, 14(1), 89. https://doi.org/10.3390/v14010089