
The Novel hDHODH Inhibitor MEDS433 Prevents Influenza Virus Replication by Blocking Pyrimidine Biosynthesis

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cells and Viruses
2.3. Cytotoxicity Assay
2.4. Antiviral Assays
2.5. Immunoblotting
2.6. hDHODH Gene Silencing
2.7. Data Analysis
3. Results
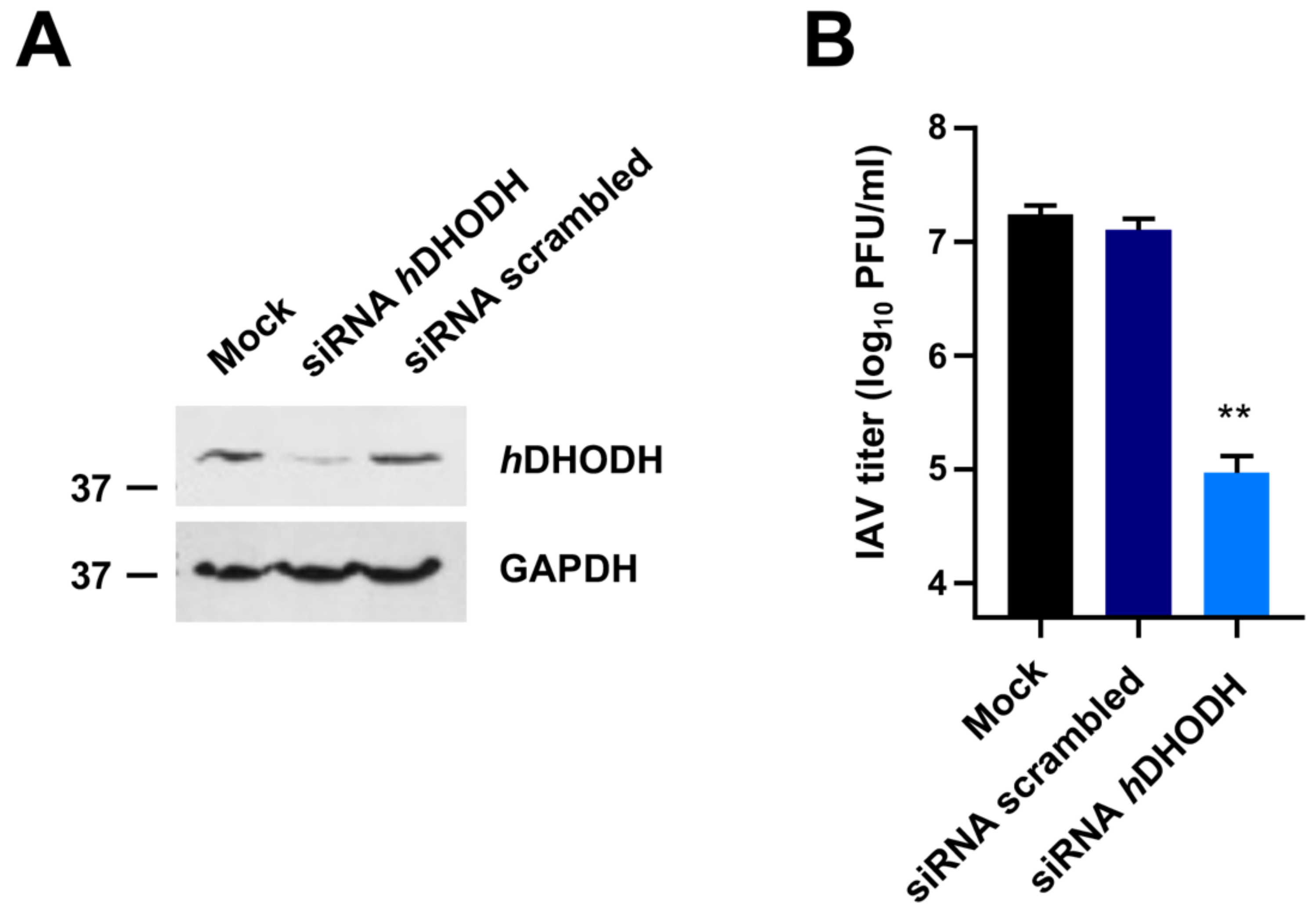
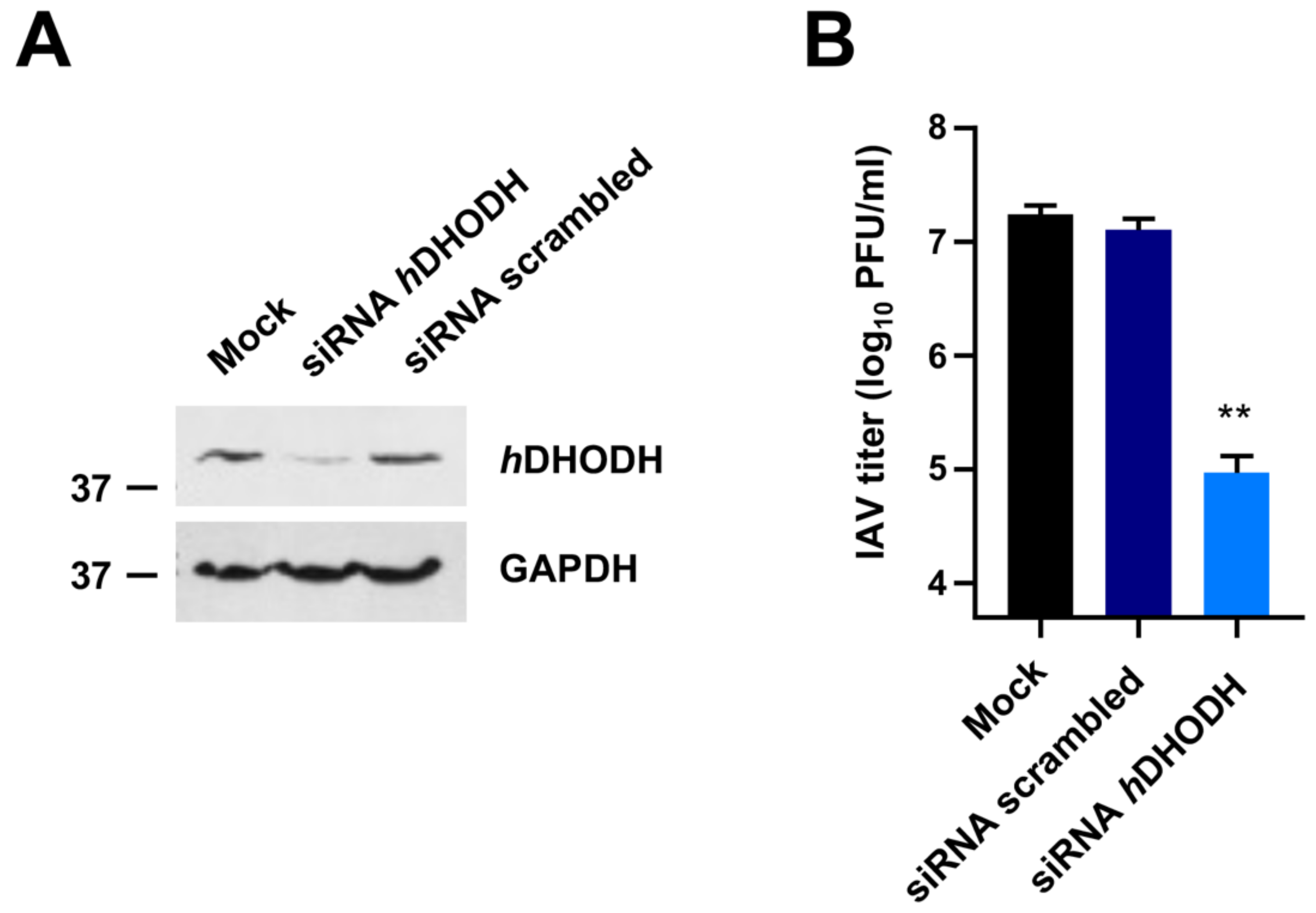
3.1. hDHODH Expression Is Required for Efficient IAV Replication
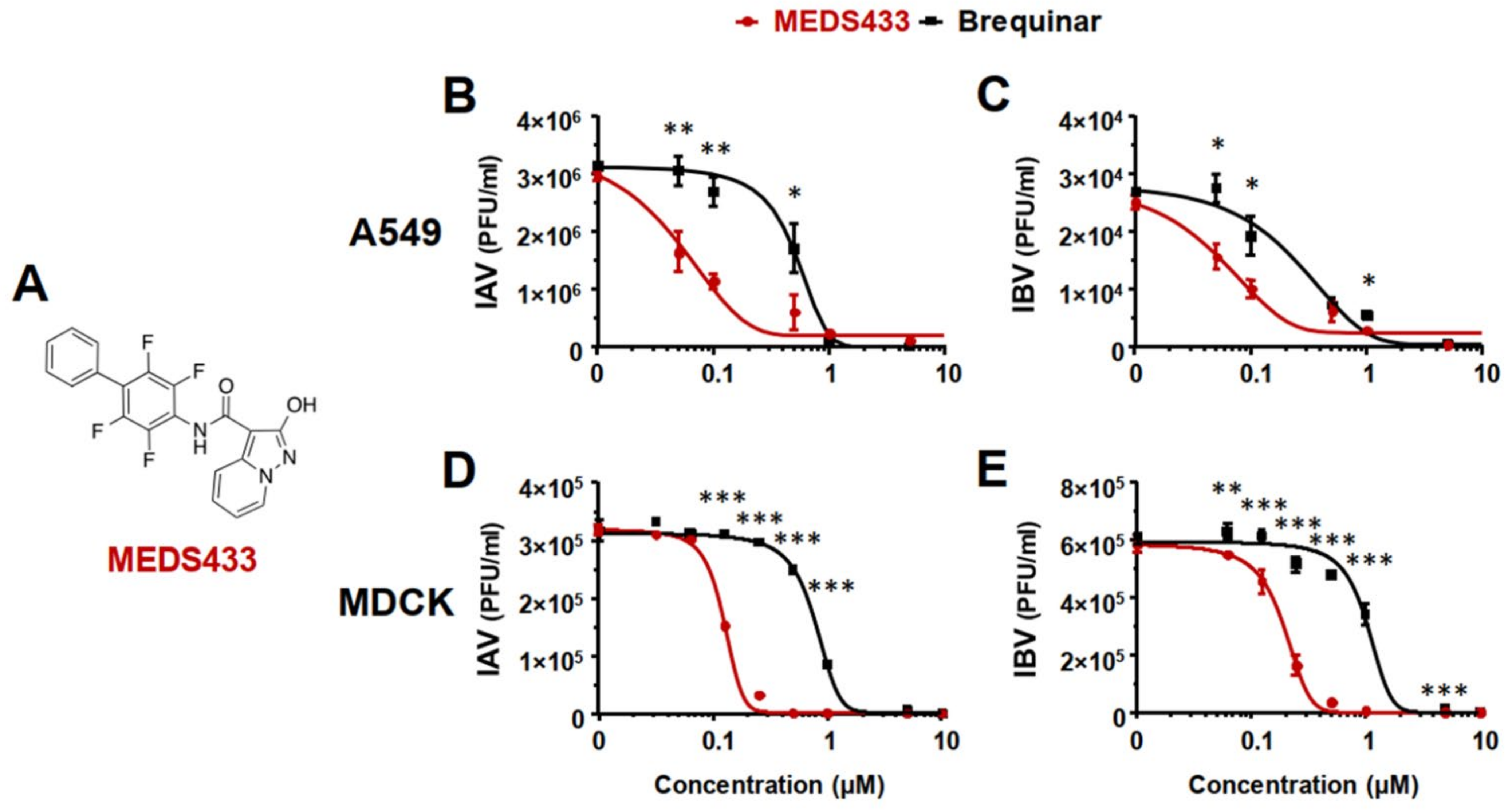
3.2. MEDS433 Inhibits IAV and IBV In Vitro Replication
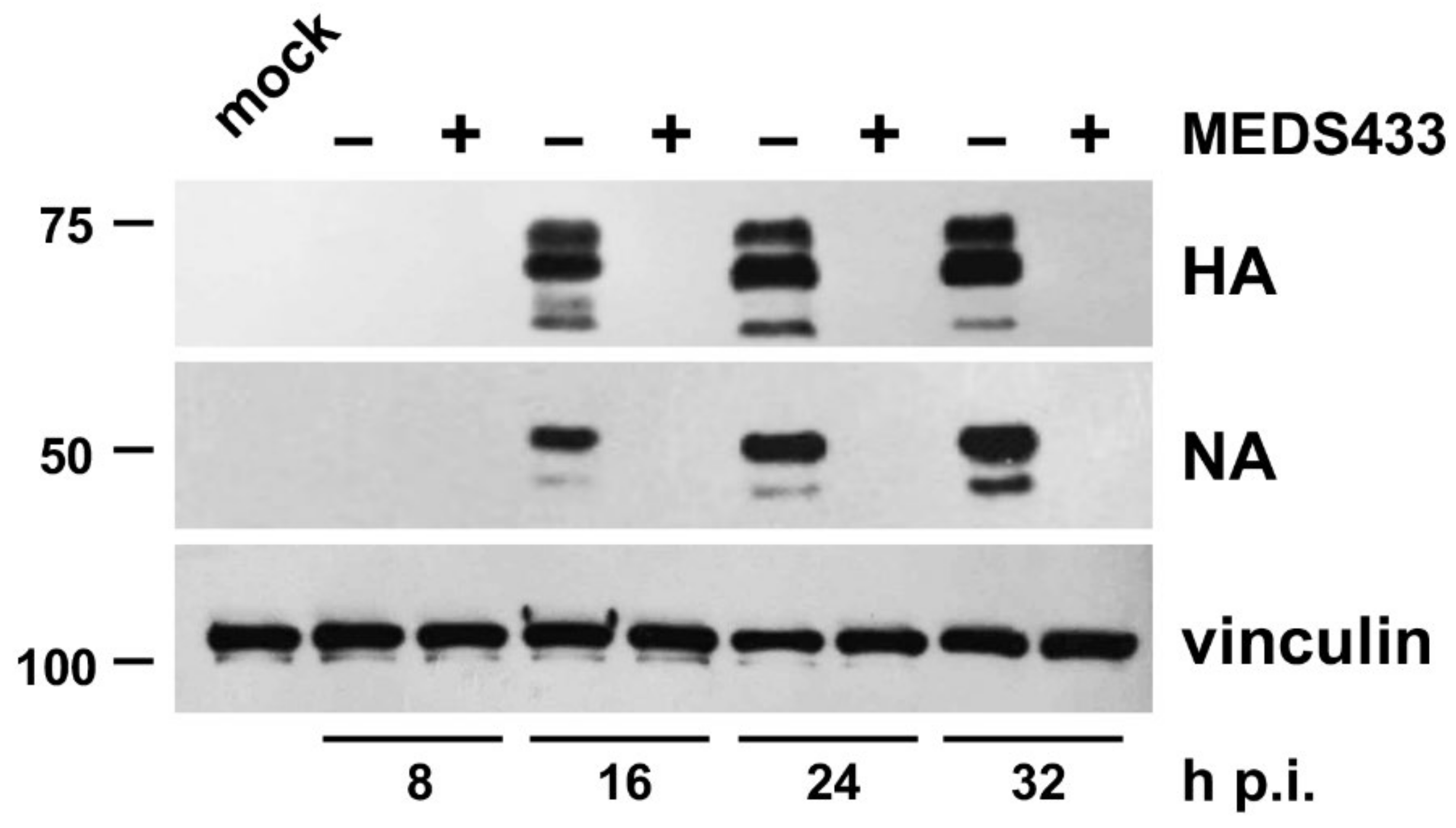
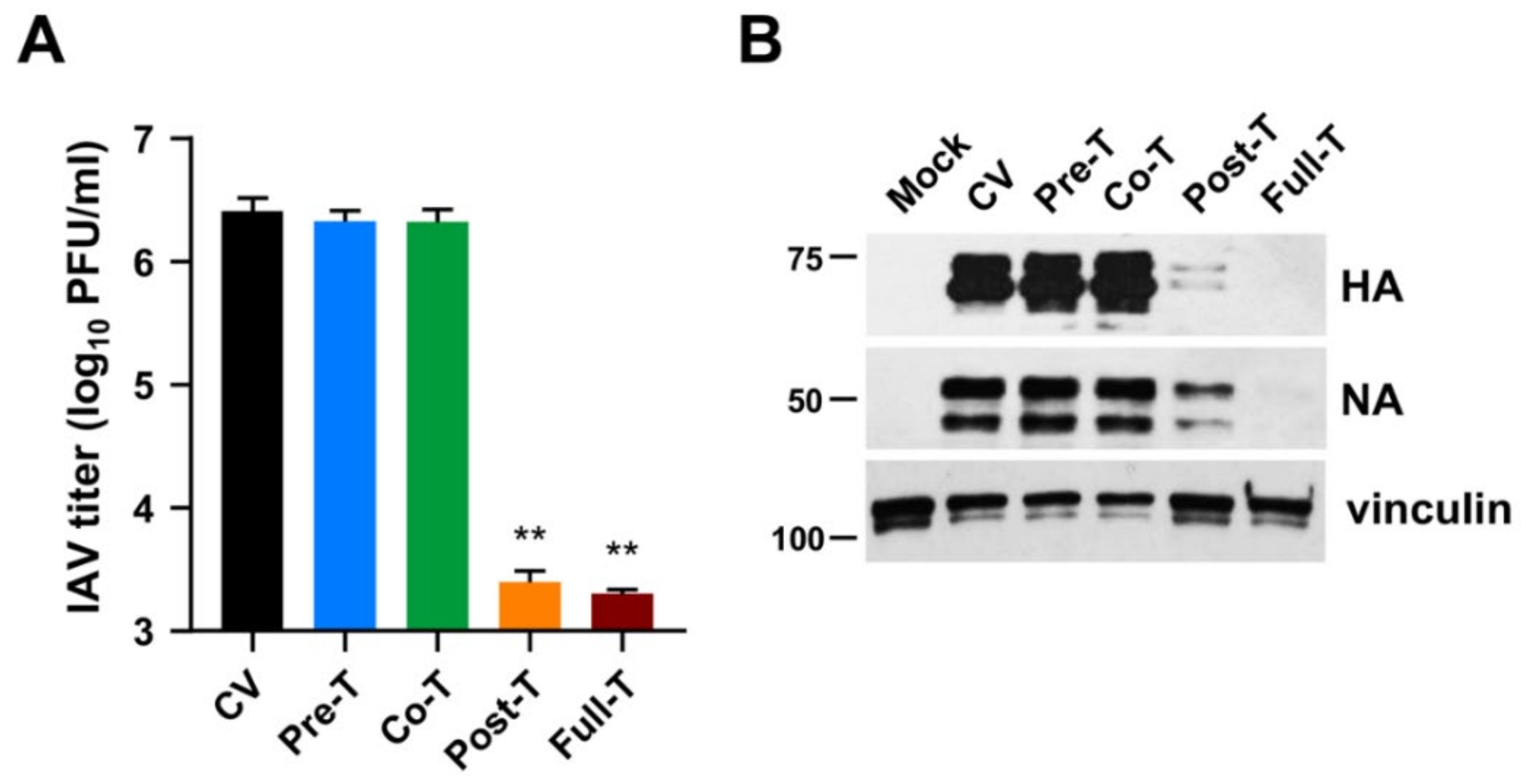
3.3. MEDS433 Affects IAV Protein Expression by Targeting a Post-Entry Phase of the Virus Replicative Cycle
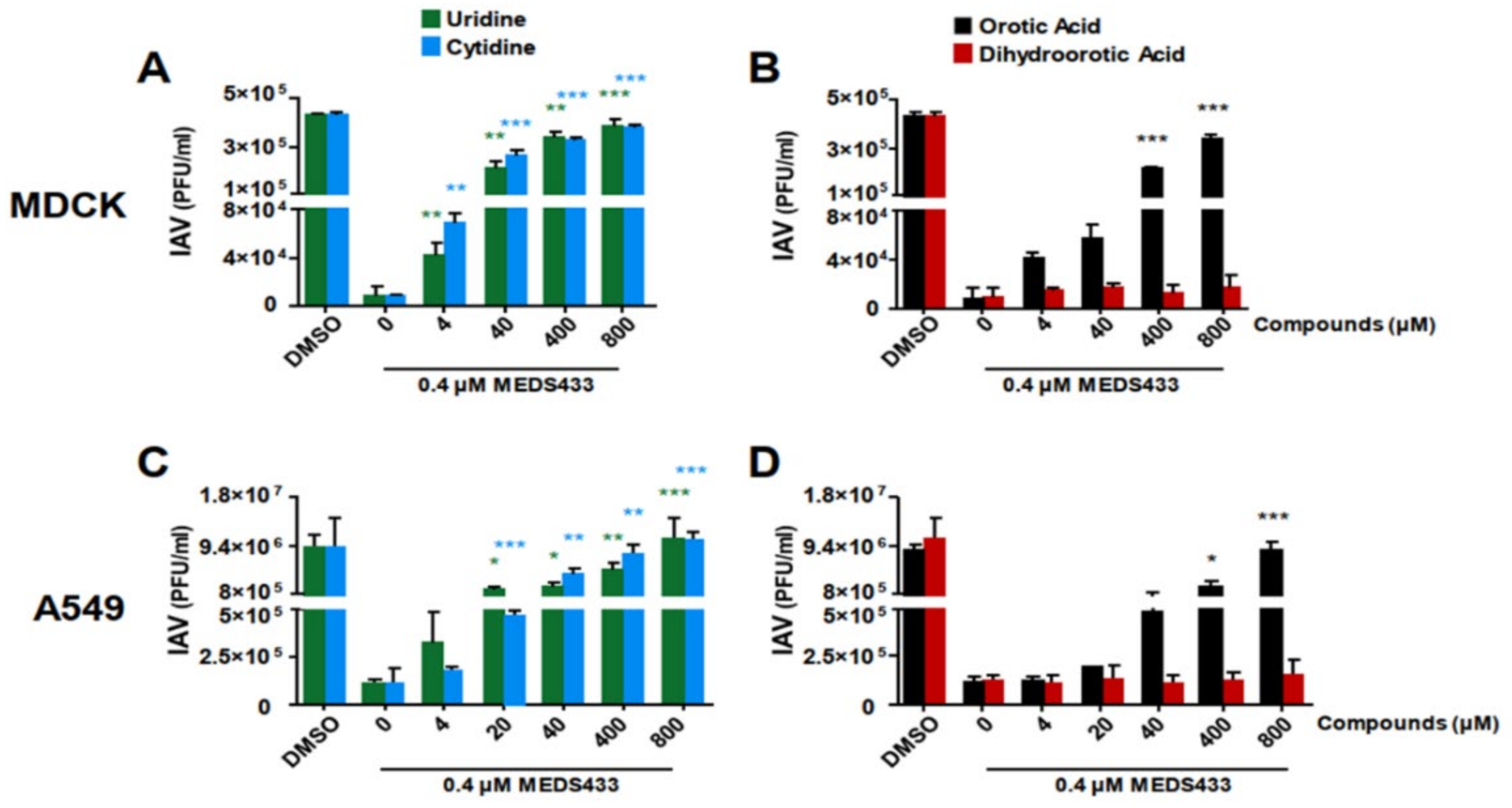
3.4. The Pyrimidine Biosynthesis Pathway in Implicated in the Anti-IV Activity of MEDS433
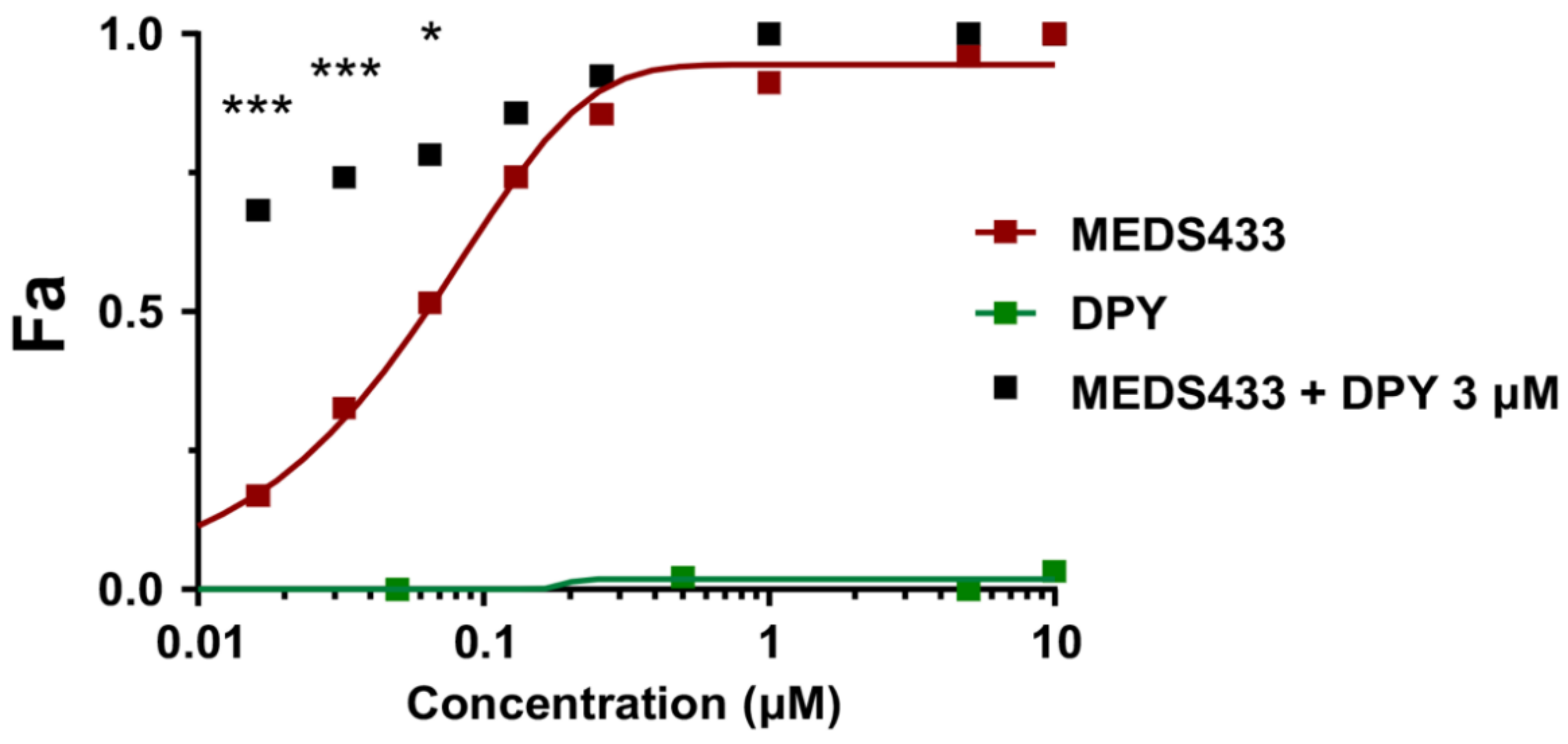
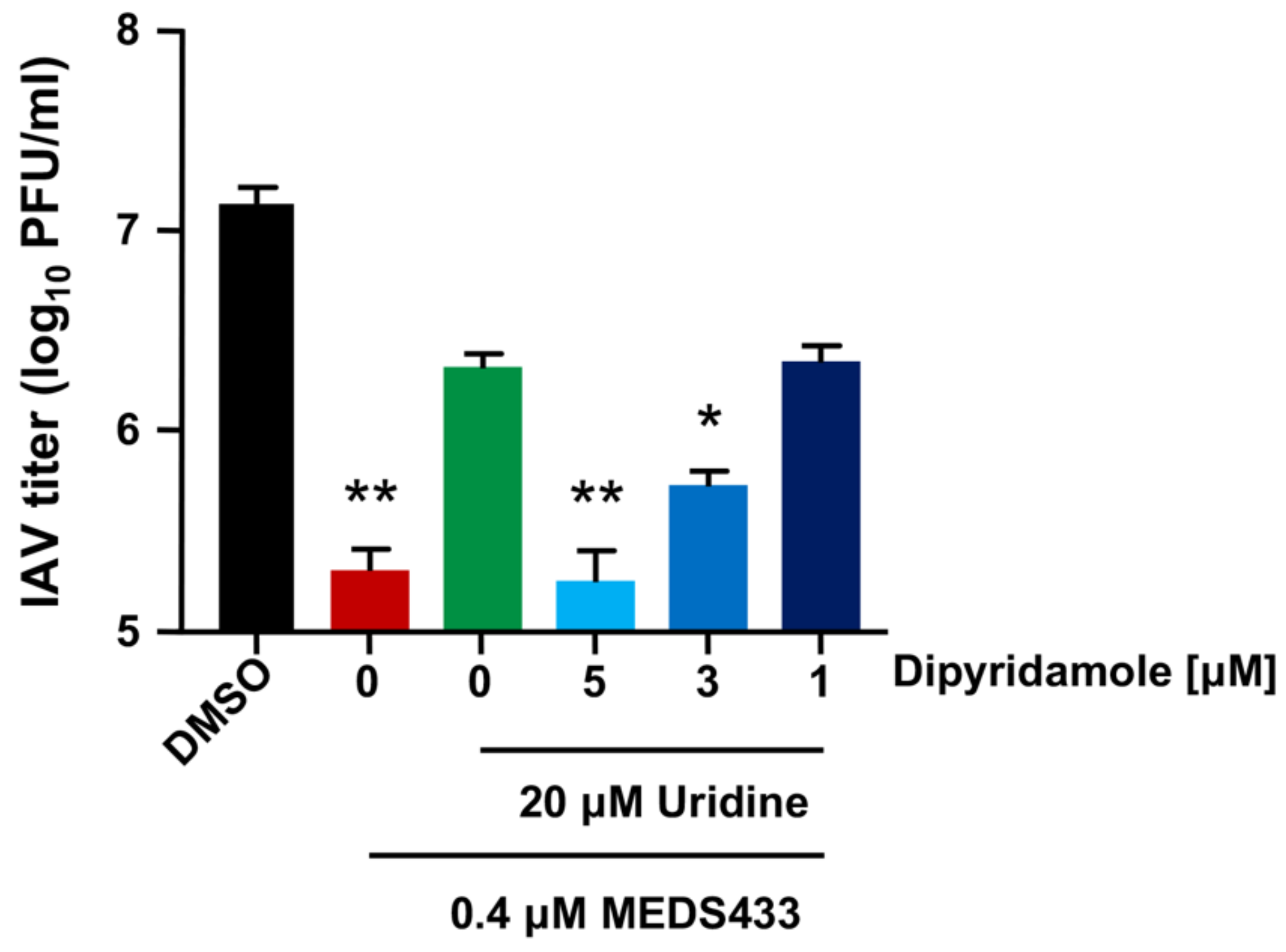
3.5. The Combination of MEDS433 with an Inhibitor of the Nucleoside Salvage Pathway Enhances the Anti-IAV Activity of the hDHODH Inhibitor
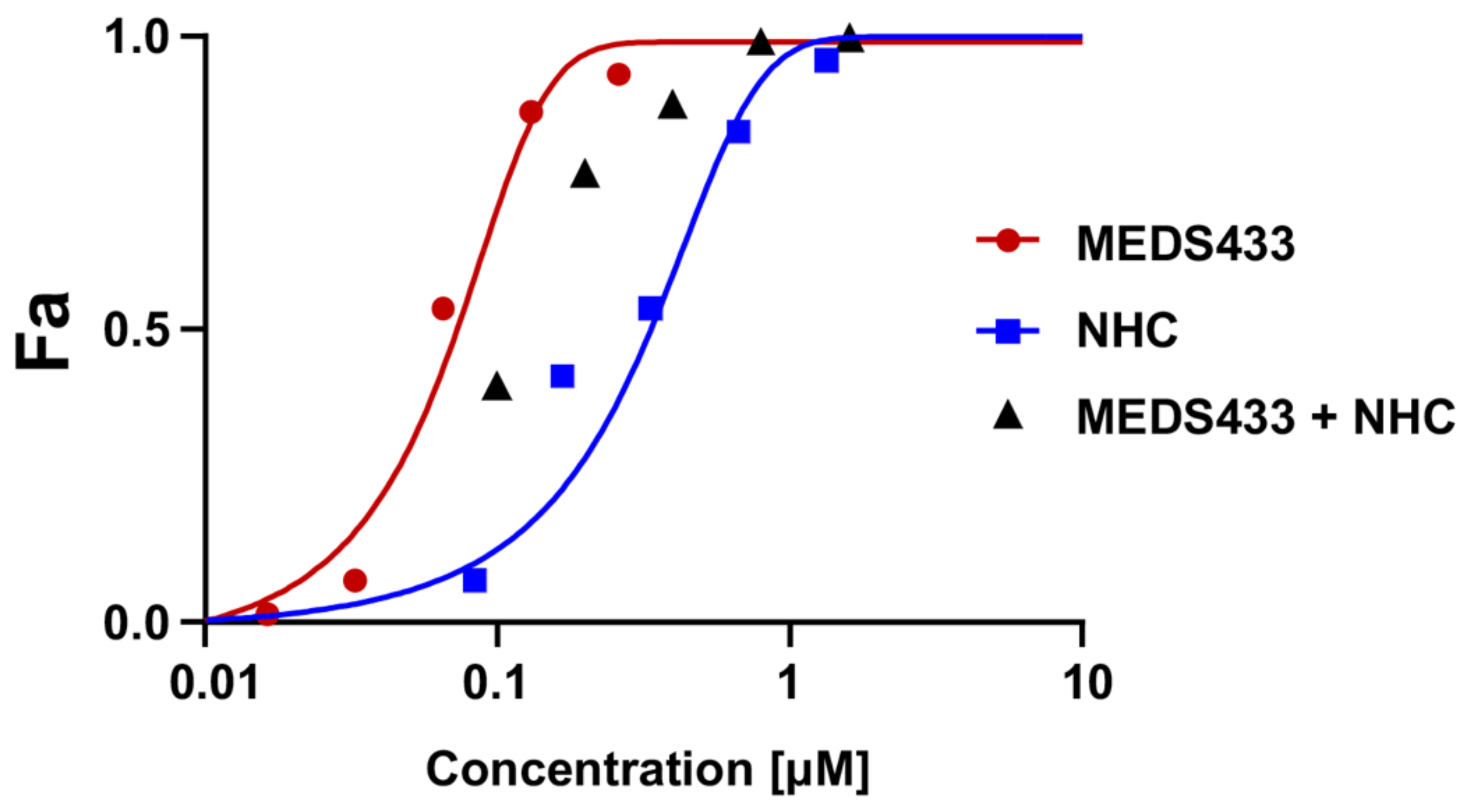
3.6. MEDS433 and the Ribonucleoside Analogue N4-Hydroxycytidine Synergistically Act against IAV Replication
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Influenza Strategy 2019–2030; WHO: Geneva, Switzerland, 2019.
- Peteranderl, C.; Herold, S.; Schmoldt, C. Human influenza virus infections. Semin. Respir. Crit. Care Med. 2016, 37, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.; Subbarao, K. Influenza. Lancet 2017, 390, 687–708. [Google Scholar] [CrossRef]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Kawaoka, T. Current and future influenza vaccines. Nat. Med. 2019, 25, 212–220. [Google Scholar] [CrossRef]
- Nachbagauer, R.; Palese, P. Is a universal influenza virus vaccine possible? Annu. Rev. Med. 2020, 71, 315–327. [Google Scholar] [CrossRef]
- Chow, E.J.; Doyle, J.D.; Uyeki, T.M. Influenza virus-related critical illness: Prevention, diagnosis, treatment. Crit. Care 2019, 23, 214. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Ramphul, K. Amantadine; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- McKimm-Breschkin, J.L. Influenza neuraminidase inhibitors: Antiviral action and mechanisms of resistance. Influenza Respir. Viruses 2013, 7, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Lampejo, T. Influenza and antiviral resistance: An overview. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1201–1208. [Google Scholar] [CrossRef]
- Govorkova, E.A.; Takashita, E.; Daniels, R.S.; Fujisaki, S.; Presser, L.D.; Patel, M.C.; Huang, W.; Lackenby, A.; Nguyen, H.T.; Pereyaslov, D.; et al. Global update on the susceptibilities of human influenza viruses to neuraminidase inhibitors and the Cap-dependent endonuclease inhibitor baloxavir, 2018–2020. Antivir. Res. 2022, 200, 105281. [Google Scholar] [CrossRef]
- Davidson, S. Treating influenza infection, from now and into the future. Front. Immunol. 2018, 9, 1946. [Google Scholar] [CrossRef]
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; De Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef]
- Shiraki, K.; Daikoku, T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol. Ther. 2020, 209, 107512. [Google Scholar] [CrossRef]
- Toots, M.; Plemper, R.K. Next-generation direct-acting influenza therapeutics. Transl. Res. 2020, 220, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Van de Wakker, S.I.; Fischer, M.J.E.; Oosting, R.S. New drug strategies to tackle viral-host interactions for the treatment of influenza virus infections. Eur. J. Pharmacol. 2017, 809, 178–190. [Google Scholar] [CrossRef]
- Okesli, A.; Khosla, C.; Bassik, M.C. Human pyrimidine nucleotide biosynthesis as a target for antiviral chemotherapy. Curr. Op. Biotech. 2017, 48, 127–134. [Google Scholar] [CrossRef]
- Gribaudo, G.; Riera, L.; Rudge, T.L.; Caposio, P.; Johnson, L.F.; Landolfo, S. Human cytomegalovirus infection induces cellular thymidylate synthase gene expression in quiescent fibroblasts. J. Gen. Virol. 2002, 83, 2983–2993. [Google Scholar] [CrossRef]
- Gribaudo, G.; Riera, L.; Caposio, P.; Maley, F.; Landolfo, S. Human cytomegalovirus requires cellular deoxycytidylate deaminase for replication in quiescent cells. J. Gen. Virol. 2003, 84, 1437–1441. [Google Scholar] [CrossRef]
- Munger, J.; Bennett, B.; Parikh, A.; Feng, X.-J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Karimi, Z.; Oskouie, A.A.; Rezaie, F.; Ajaminejad, F.; Marashi, S.M.; Azad, T.-M. The effect of influenza virus on the metabolism of peripheral blood mononuclear cells with a metabolomics approach. J. Med. Virol. 2022, 94, 4383–4392. [Google Scholar] [CrossRef]
- Reis, R.A.G.; Calil, F.A.; Feliciano, P.R.; Pinheiro, M.P.; Nonato, M.C. The dihydroorotate dehydrogenases: Past and present. Arch. Biochem. Biophys. 2017, 632, 75–191. [Google Scholar] [CrossRef]
- Loeffler, M.; Carrey, E.A.; Knecht, W. The pathway to pyrimidines: The essential focus on dihydroorotate dehydrogenase, the mitochondrial enzyme coupled to the respiratory chain. Nucleosides Nucleotides Nucleic Acids 2020, 11, 1–25. [Google Scholar] [CrossRef]
- Sainas, S.; Pippione, A.C.; Lupino, E.; Giorgis, M.; Circosta, P.; Gaidano, V.; Goyal, P.; Bonanni, D.; Rolando, B.; Cignetti, A.; et al. Targeting myeloid differentiation using potent 2-Hydroxypyrazolo [1,5-α] pyridine scaffold-based human dihydroorotate dehydrogenase inhibitors. J. Med. Chem. 2018, 61, 6034–6055. [Google Scholar] [CrossRef]
- Sainas, S.; Giorgis, M.; Circosta, P.; Gaidano, V.; Bonanni, D.; Pippione, A.C.; Bagnati, R.; Passoni, A.; Qiu, Y.; Cojocaru, C.F.; et al. Targeting acute myelogenous leukemia using potent human dihydroorotate dehydrogenase inhibitors based on the 2-hydroxypyrazolo[1,5-α] pyridine scaffold: SAR of the biphenyl moiety. J. Med. Chem. 2021, 64, 5404–5428. [Google Scholar] [CrossRef]
- Sainas, S.; Giorgis, M.; Circosta, P.; Poli, G.; Alberti, M.; Passoni, A.; Gaidano, V.; Pippione, A.C.; Vitale, N.; Bonanni, D.; et al. Targeting acute myelogenous leukemia using potent human dihydroorotate dehydrogenase inhibitors based on the 2-hydroxypyrazolo[1,5-α] pyridine scaffold: SAR of the aryloxyaryl moiety. J. Med. Chem. 2022, 64, 5404–5428. [Google Scholar] [CrossRef]
- Peters, G.J. Re-evaluation of Brequinar sodium, a dihydroorotate dehydrogenase inhibitor. Nucleosides Nucleotides Nucleic Acids 2018, 37, 666–678. [Google Scholar] [CrossRef]
- Park, J.-G.; Ávila-Pérez, G.; Nogales, A.; Blanco-Lobo, P.; De la Torre, J.C.; Martínez-Sobrido, L. Identification and characterization of novel compounds with broad-spectrum antiviral activity against influenza A and B viruses. J. Virol. 2020, 94, e02149-e19. [Google Scholar] [CrossRef] [Green Version]
- Boschi, D.; Pippione, A.C.; Sainas, S.; Lolli, M.L. Dihydroorotate dehydrogenase inhibitors in anti-infective drug research. Eur. J. Med. Chem. 2019, 183, 111681. [Google Scholar] [CrossRef]
- Luganini, A.; Sibille, G.; Mognetti, B.; Sainas, S.; Pippione, A.C.; Giorgis, M.; Boschi, D.; Lolli, M.L.; Gribaudo, G. Effective deploying of a novel DHODH inhibitor against herpes simplex type 1 and type 2 replication. Antivir. Res. 2021, 189, 105057. [Google Scholar] [CrossRef]
- Calistri, A.; Luganini, A.; Mognetti, B.; Elder, E.; Sibille, G.; Conciatori, V.; Del Vecchio, C.; Sainas, S.; Boschi, D.; Montserrat, N.; et al. The new generation hDHODH inhibitor MEDS433 hinders the in vitro replication of SARS-CoV-2 and other human coronaviruses. Microorganisms 2021, 9, 1731. [Google Scholar] [CrossRef]
- Luganini, A.; Terlizzi, M.E.; Catucci, G.; Gilardi, G.; Maffei, M.E.; Gribaudo, G. The cranberry extract Oximacro exerts in vitro virucidal activity against influenza virus by interfering with hemagglutinin. Front. Microbiol. 2018, 9, 1826. [Google Scholar] [CrossRef]
- Simon, L.M.; Morandi, E.; Luganini, A.; Gribaudo, G.; Martinez-Sobrido, L.; Turner, D.H.; Oliviero, S.; Incarnato, D. In vivo analysis of influenza A mRNA secondary structures identifies critical regulatory motifs. Nucleic Acids Res. 2019, 47, 7003–7017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauwels, R.; Balzarini, J.; Baba, M.; Snoeck, R.; Schols, D.; Herdewijn, P.; Desmyter, J.; DeClercq, E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 1988, 20, 309–321. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Martin, N. CompuSyn for Drug Combinations: PC Software and User’s Guide—A Computer Program for Quantitation of Synergism and Antagonism in Drug Combinations, and the Determination of IC50 and ED50 and LD50 Bvalues; ComboSyn: Paramus, NJ, USA, 2005. [Google Scholar]
- Luganini, A.; Caposio, P.; Landolfo, S.; Gribaudo, G. Phosphorothioate-modified oligodeoxynucleotides inhibit human cytomegalovirus replication by blocking virus entry. Antimicrob. Agents Chemother. 2008, 52, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, G.A. Dipyridamole. N. Engl. J. Med. 1987, 316, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Schaper, W. Dipyridamole, an underestimated vascular protective drug. Cardiovasc. Drugs Ther. 2005, 19, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.L.; Sherali, A.; Mo, Z.P.; Tse, C.M. Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells: ENT2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J. Biol. Chem. 2000, 275, 8375–8381. [Google Scholar] [CrossRef] [Green Version]
- Pizzorno, G.; Cao, D.; Leffert, J.J.; Russell, R.L.; Zhang, D.; Handschumacher, R.E. Homeostatic control of uridine and the role of uridine phosphorylase: A biological and clinical update. Biochim. Biophys. Acta 2002, 1587, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.-J.; Toots, M.; Lee, S.; Lee, M.-E.; Ludeke, B.; Luczo, J.M.; Ganti, K.; Cox, R.M.; Sticher, Z.M.; Edpuganti, V.; et al. Orally efficacious broad-spectrum ribonucleoside analog inhibitor of influenza and respiratory syncytial viruses. Antimicrob. Agents Chemother. 2018, 62, e00766-e18. [Google Scholar] [CrossRef] [Green Version]
- Toots, M.; Yoon, J.-J.; Cox, R.M.; Hart, M.; Sticher, Z.M.; Makhsous, N.; Plesker, R.; Barrena, A.H.; Reddy, P.G.; Mitchell, D.G.; et al. Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci. Transl. Med. 2019, 11, eaax5866. [Google Scholar] [CrossRef]
- Amesh Adalja, A.; Inglesby, T. Broad-spectrum antiviral agents: A crucial pandemic tool. Exp. Rev. Anti-Infect. Ther. 2019, 17, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Coehlo, A.R.; Oliveira, P.J. Dihydroorotate dehydrogenase inhibitors in SARS-CoV-2 infection. Eur. J. Clin. Investig. 2020, 50, e13366. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, S.; Song, K.; Ye, J.; Li, W.; Zhong, Y.; Feng, Z.; Liang, S.; Cai, Z.; Xu, K. A broad antiviral strategy: Inhibitors of human DHODH pave the way for host-targeting antivirals against emerging and re-emerging Viruses. Viruses 2022, 14, 928. [Google Scholar] [CrossRef]
- Cheung, N.N.; Lai, K.K.; Dai, J.; Kok, K.H.; Chen, H.; Chan, K.-H.; Yuen, K.-Y.; Tsun Sao, R.Y. Broad-spectrum inhibition of common respiratory RNA viruses by a pyrimidine synthesis inhibitor with involvement of the host antiviral response. J. Gen. Virol. 2017, 98, 946–954. [Google Scholar] [CrossRef]
- Xiong, R.; Zhang, L.; Li, S.; Sun, Y.; Ding, M.; Wang, Y.; Zhao, Y.; Wu, Y.; Shang, W.; Jiang, X.; et al. Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly emerged coronavirus SARS-CoV-2. Protein Cell 2020, 11, 723–739. [Google Scholar] [CrossRef]
- Gradl, S.N.; Mueller, T.; Ferrara, S.; Sheikh, S.E.; Janzer, A.; Zhou, H.-J.; Friberg, A.; Guenther, J.; Schaefer, M.; Stellfeld, T.; et al. Discovery of BAY 2402234 by phenotypic screening: A human dihydroorotate dehydrogenase (DHODH) inhibitor in clinical trials for the treatment of myeloid malignancies. In Proceedings of the American Association for Cancer Research Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019; Volume 79 13 Suppl. Abstract 2. [Google Scholar]
- Wang, Q.Y.; Bushell, S.; Qing, M.; Xu, H.Y.; Bonavia, A.; Nunes, S.; Zhou, J.; Poh, M.K.; Florez de Sessions, P.; Niyomrattanakit, P.; et al. Inhibition of dengue virus through suppression of host pyrimidine biosynthesis. J. Virol. 2011, 85, 6548–6556. [Google Scholar] [CrossRef] [Green Version]
- Smee, D.F.; Hurst, B.L.; Day, C.W. D282, a non-nucleoside inhibitor of influenza virus infection that interferes with de novo pyrimidine biosynthesis. Antivir. Chem. Chemother. 2012, 22, 263–272. [Google Scholar] [CrossRef]
- Grandin, C.; Lucas-Hourani, M.; Janin, Y.L.; Dauzonne, D.; Munier-Lehmann, H.; Paturet, A.; Taborik, F.; Vabret, A.; Contamin, H.; Tangy, F.; et al. Respiratory syncytial virus infection in macaques is not suppressed by intranasal sprays of pyrimidine biosynthesis inhibitors. Antiviral Res. 2016, 125, 58–62. [Google Scholar] [CrossRef]
- Gaidano, V.; Houshmand, M.; Vitale, N.; Carr, G.; Morotti, A.; Tenace, V.; Rapelli, S.; Sainas, S.; Pippione, A.C.; Giorgis, M.; et al. The synergism between DHODH inhibitors and dipyridamole leads to metabolic lethality in acute myeloid leukemia. Cancers 2021, 13, 1003. [Google Scholar] [CrossRef]
- Gregov, D.; Jenkins, A.; Duncan, E.; Sieber, D.; Rodgers, S.; Duncan, B.; Bochner, F.; Lloyd, J. Dipyridamole: Pharmacokinetics and effects on aspects of platelet function in man. Br. J. Clin. Pharmacol. Soc. 1987, 24, 425–434. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martín-Quirós, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Hobartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Stegmann, K.M.; Dickmanns, A.; Heinen, N.; Blaurock, C.; Karrasch, T.; Breithaupt, A.; Klopfleisch, R.; Uhlig, N.; Eberlein, V.; Issmail, L.; et al. Inhibitors of dihydroorotate dehydrogenase cooperate with molnupiravir and N4-hydroxycytidine to suppress SARS-CoV-2 replication. iScience 2022, 25, 104293. [Google Scholar] [CrossRef]
- Schultz, D.C.; Johnson, R.M.; Ayyanathan, K.; Miller, J.; Whig, K.; Kamalia, B.; Dittmar, M.; Weston, S.; Hammond, H.L.; Dillen, C.; et al. Pyrimidine inhibitors synergize with nucleoside analogues to block SARS-CoV-2. Nature 2022, 604, 134–140. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IV | Cell Line | EC50 (μM) a | EC90 (μM) b | CC50 (μM) c | SI d |
|---|---|---|---|---|---|
| IAV | A549 | 0.064 ± 0.01 μM | 0.264 ± 0.002 μM | 64.25 ± 3.12 μΜ | 1104 |
| Calu-3 | 0.055 ± 0.003 μΜ | 0.675 ± 0.05 μΜ | 54.67 ± 3.86 μΜ | 994 | |
| MDCK | 0.141 ± 0.021 μM | 0.256 ± 0.052 μΜ | 119.8 ± 6.21 μΜ | 850 | |
| IBV | A549 | 0.065 ± 0.005 μΜ | 0.365 ± 0.09 μΜ | 64.25 ± 3.12 μΜ | 988 |
| Calu-3 | 0.052 ± 0.006 μΜ | 0.807 ± 0.08 μΜ | 54.67 ± 3.86 μΜ | 1051 | |
| MDCK | 0.170 ± 0.019 μM | 0.330 ± 0.013 μΜ | 119.8 ± 6.21 μΜ | 705 |
| MEDS433 Concentration (Fold of EC50 a) + DPY 3 μM | MEDS433/DPY CI b | Drug Combination Effect c of MEDS433 and DPY |
|---|---|---|
| 4× | 0.824 ± 0.023 | Moderate Synergism |
| 2× | 0.607 ± 0.116 | Synergism |
| 1× | 0.341 ± 0.025 | Synergism |
| 0.5× | 0.203 ± 0.044 | Strong Synergism |
| 0.25× | 0.126 ± 0.032 | Strong Synergism |
| MEDS433/NHC Combination at Equipotent Ratio (fold of EC50 a) | MEDS433/NHC CI b | Drug Combination Effect c of MEDS433 and NHC |
|---|---|---|
| 4× | 0.075 ± 0.004 | Very Strong Synergism |
| 2× | 0.484 ± 0.011 | Synergism |
| 1× | 0.825 ± 0.004 | Moderate Synergism |
| 0.5× | 0.604 ± 0.028 | Synergism |
| 0.25× | 0.625 ± 0.019 | Synergism |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sibille, G.; Luganini, A.; Sainas, S.; Boschi, D.; Lolli, M.L.; Gribaudo, G. The Novel hDHODH Inhibitor MEDS433 Prevents Influenza Virus Replication by Blocking Pyrimidine Biosynthesis. Viruses 2022, 14, 2281. https://doi.org/10.3390/v14102281
Sibille G, Luganini A, Sainas S, Boschi D, Lolli ML, Gribaudo G. The Novel hDHODH Inhibitor MEDS433 Prevents Influenza Virus Replication by Blocking Pyrimidine Biosynthesis. Viruses. 2022; 14(10):2281. https://doi.org/10.3390/v14102281
Chicago/Turabian StyleSibille, Giulia, Anna Luganini, Stefano Sainas, Donatella Boschi, Marco Lucio Lolli, and Giorgio Gribaudo. 2022. "The Novel hDHODH Inhibitor MEDS433 Prevents Influenza Virus Replication by Blocking Pyrimidine Biosynthesis" Viruses 14, no. 10: 2281. https://doi.org/10.3390/v14102281
APA StyleSibille, G., Luganini, A., Sainas, S., Boschi, D., Lolli, M. L., & Gribaudo, G. (2022). The Novel hDHODH Inhibitor MEDS433 Prevents Influenza Virus Replication by Blocking Pyrimidine Biosynthesis. Viruses, 14(10), 2281. https://doi.org/10.3390/v14102281