

How EBV Infects: The Tropism and Underlying Molecular Mechanism for Viral Infection


Abstract
1. Introduction
2. EBV Tropism and Related Diseases
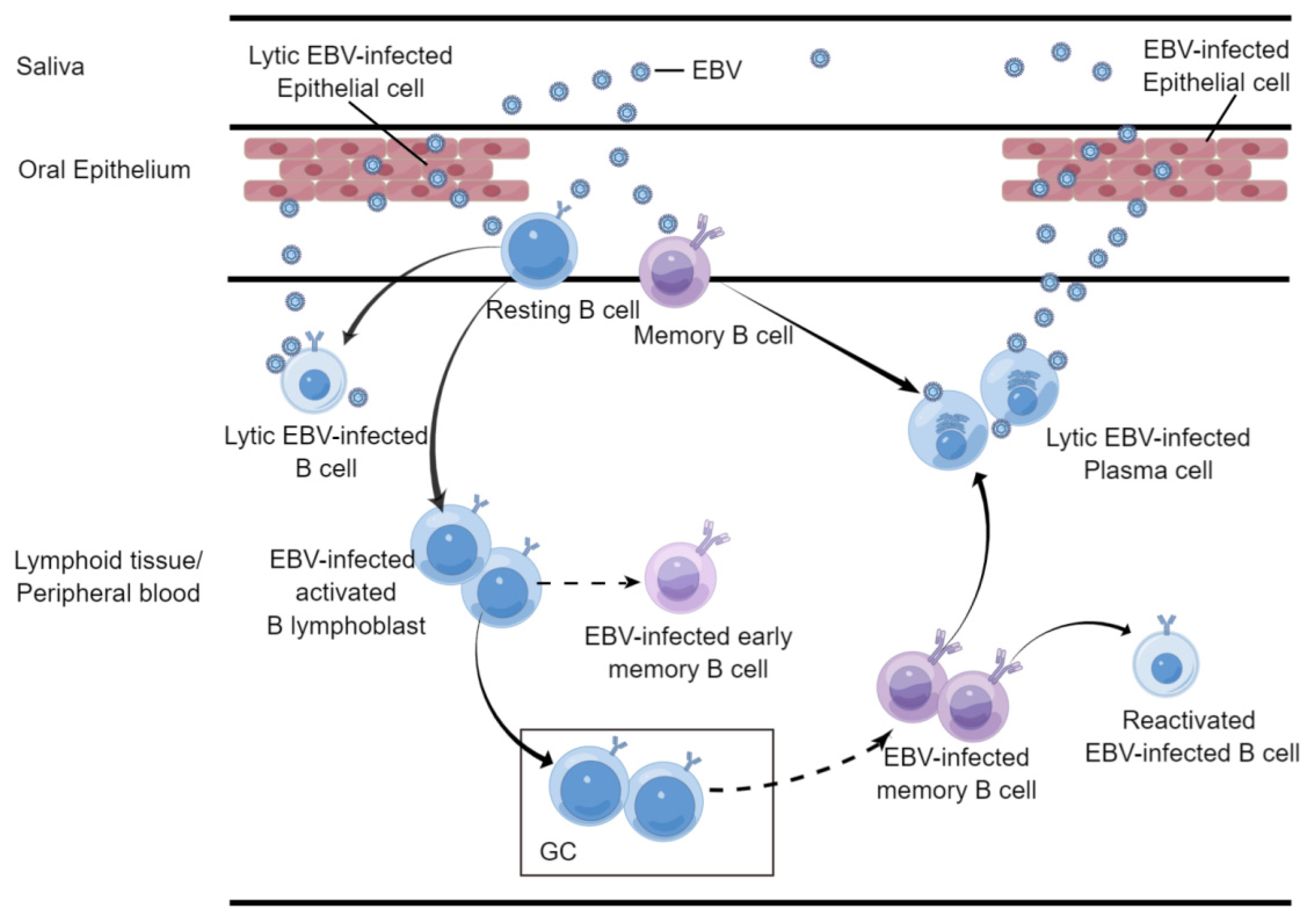
2.1. B Cell
2.2. Epithelial Cell
2.3. NK/T Cell
2.4. Others
3. EBV Entry into Target Cells
3.1. Attachment
3.2. Binding and Entry
3.3. Membrane Fusion
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein- Barr Virus and Cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- WHO; International Agency for Research on Cancer. Epstein-Barr Virus and Kaposi’s Sarcoma Herpesvirus/Human Herpesvirus 8: IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 1997; Volume 70, 492p.
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agents Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Houen, G.; Trier, N.H. Epstein-Barr Virus and Systemic Autoimmune Diseases. Front. Immunol. 2021, 11, 587380. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Steinman, L. Epstein-Barr virus and multiple sclerosis. Science 2022, 375, 264–265. [Google Scholar] [CrossRef]
- Baer, R.; Bankier, A.T.; Biggin, M.D.; Deininger, P.L.; Farrell, P.J.; Gibson, T.J.; Hatfull, G.; Hudson, G.S.; Satchwell, S.C.; Seguin, C. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 1984, 310, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Notarte, K.I.; Senanayake, S.; Macaranas, I.; Albano, P.M.; Mundo, L.; Fennell, E.; Leoncini, L.; Murray, P. MicroRNA and Other Non-Coding RNAs in Epstein-Barr Virus-Associated Cancers. Cancers 2021, 13, 3909. [Google Scholar] [CrossRef]
- Young, L.S.; Arrand, J.R.; Murray, P.G. EBV gene expression and regulation. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; Volume 27, pp. 461–489. [Google Scholar]
- Moehl, B.S.; Chen, J.; Sathiyamoorthy, K.; Jardetzky, T.S.; Longnecker, R. Structural and Mechanistic Insights into the Tropism of Epstein-Barr Virus. Mol. Cells 2016, 39, 286–291. [Google Scholar] [CrossRef]
- Moehl, B.S.; Chen, J.; Longnecker, R. Gammaherpesvirus entry and fusion: A tale how two human pathogenic viruses enter their host cells. Adv. Virus Res. 2019, 104, 313–343. [Google Scholar] [CrossRef]
- Chen, J.; Longnecker, R. Epithelial cell infection by Epstein-Barr virus. FEMS Microbiol. Rev. 2019, 43, 674–683. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L.M. EBV glycoproteins: Where are we now? Future Virol. 2015, 10, 1155–1162. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar] [CrossRef]
- Sathiyamoorthy, K.; Hu, Y.X.; Mohl, B.S.; Chen, J.; Longnecker, R.; Jardetzky, T.S. Structural basis for Epstein-Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nat. Commun. 2016, 7, 13557. [Google Scholar] [CrossRef]
- Chiu, Y.-F.; Sugden, B. Epstein-Barr Virus: The Path from Latent to Productive Infection. Annu. Rev. Virol. 2016, 3, 359–372. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L.M. Epstein-Barr virus replicating in epithelial cells. Proc. Natl. Acad. Sci. USA 2014, 111, 16242–16243. [Google Scholar] [CrossRef]
- Sausen, D.G.; Bhutta, M.S.; Gallo, E.S.; Dahari, H.; Borenstein, R. Stress-Induced Epstein-Barr Virus Reactivation. Biomolecules 2021, 11, 1380. [Google Scholar] [CrossRef]
- Pope, J.H. Establishment of cell lines from peripheral leucocytes in infectious mononucleosis. Nature 1967, 216, 810–811. [Google Scholar] [CrossRef]
- Pope, J.H.; Horne, M.K.; Scott, W. Transformation of foetal human keukocytes in vitro by filtrates of a human leukaemic cell line containing herpes-like virus. Int. J. Cancer 1968, 3, 857–866. [Google Scholar] [CrossRef]
- Horley-Lawson, D.A. EBV Persistence-Introducing the Virus. In Current Topics in Microbiology and Immunology; Arber, W., Braun, W., Haas, R., Henle, W., Hofschmeider, P.H., Jerne, N.K., Koldovsky, P., Koprowski, H., Maalow, O., Rott, R., et al., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 390, pp. 151–209. [Google Scholar] [CrossRef]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef]
- Jiang, R.; Scott, R.S.; Hutt-Fletcher, L.M. Epstein-Barr virus shed in saliva is high in B-cell-tropic glycoprotein gp42. J. Virol. 2006, 80, 7281–7283. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Herrera, R.; Palefsky, J.M. Epstein-Barr Virus Transcytosis through Polarized Oral Epithelial Cells. J. Virol. 2013, 87, 8179–8194. [Google Scholar] [CrossRef]
- Grimm, J.M.; Schmeling, D.O.; Dunmire, S.K.; Knight, J.A.; Mullan, B.D.; Ed, J.A.; Brundage, R.C.; Hogquist, K.A.; Balfour, H.H., Jr. Prospective studies of infectious mononucleosis in university students. Clin. Transl. Immunol. 2016, 5, e94. [Google Scholar] [CrossRef]
- Hadinoto, V.; Shapiro, M.; Sun, C.C.; Thorley-Lawson, D.A. The Dynamics of EBV Shedding Implicate a Central Role for Epithelial Cells in Amplifying Viral Output. PLoS Pathog. 2009, 5, e1000496. [Google Scholar] [CrossRef]
- Niederman, J.C.; Miller, G.; Pearson, H.A.; Pagano, J.S.; Dowaliby, J.M. Infectious mononucleosis. Epstein-Barr-virus shedding in saliva and the oropharynx. N. Engl. J. Med. 1976, 294, 1355–1359. [Google Scholar] [CrossRef]
- Kurth, J.; Spieker, T.; Wustrow, J.; Strickler, J.G.; Hansmann, M.L.; Rajewsky, K.; Kuppers, R. EBV-infected B cells in infectious mononucleosis: Viral strategies for spreading in the B cell compartment and establishing latency. Immunity 2000, 13, 485–495. [Google Scholar] [CrossRef]
- Kurth, J.; Hansmann, M.L.; Rajewsky, K.; Kuppers, R. Epstein-Barr virus-infected B cells expanding in germinal centers of infectious mononucleosis patients do not participate in the germinal center reaction. Proc. Natl. Acad. Sci. USA 2003, 100, 4730–4735. [Google Scholar] [CrossRef]
- Kempkes, B.; Spitovsky, D.; Jansen-Durr, P.; Ellwart, J.W.; Kremmer, E.; Delecluse, H.-J.; Rottenberger, C.; Bornkamm, G.W.; Hammerschmidt, W. B-cell proliferation and induction of early G-1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J. 1995, 14, 88–96. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Mann, K.P. Early events in Epstein-Barr virus infection provide a model for B cell activation. J. Exp. Med. 1985, 162, 45–59. [Google Scholar] [CrossRef]
- Roughan, J.E.; Thorley-Lawson, D.A. The Intersection of Epstein-Barr Virus with the Germinal Center. J. Virol. 2009, 83, 3968–3976. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, D.A. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.M.; Kieff, E.D. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc. Natl. Acad. Sci. USA 1997, 94, 12592–12597. [Google Scholar] [CrossRef]
- Mosialos, G.; Birkenbach, M.; Yalamanchili, R.; Vanarsdale, T.; Kieff, C.W.; Elliott. The Epstein-Barr Virus Transforming Protein LMP1 Engages Signaling Proteins for the Tumor Necrosis Factor Receptor Family. Cell 1995, 80, 389–399. [Google Scholar] [CrossRef]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the Burkitt’s lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar] [CrossRef]
- Laichalk, L.L.; Hochberg, D.; Babcock, G.J.; Freeman, R.B.; Thorley-Lawson, D.A. The dispersal of mucosal memory B cells: Evidence from persistent EBV infection. Immunity 2002, 16, 745–754. [Google Scholar] [CrossRef]
- Crawford, D.H.; Ando, I. EB virus induction is associated with B-cell maturation. Immunology 1986, 59, 405–409. [Google Scholar]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef]
- SoRelle, E.D.; Dai, J.; Reinoso-Vizcaino, N.M.; Barry, A.P.; Chan, C.; Luftig, M.A. Time-resolved transcriptomes reveal diverse B cell fate trajectories in the early response to Epstein-Barr virus infection. Cell Rep. 2022, 40, 111286. [Google Scholar] [CrossRef] [PubMed]
- Rostgaard, K.; Balfour, H.H., Jr.; Jarrett, R.; Erikstrup, C.; Pedersen, O.; Ullum, H.; Nielsen, L.P.; Voldstedlund, M.; Hjalgrim, H. Primary Epstein-Barr virus infection with and without infectious mononucleosis. PLoS ONE 2019, 14, e0226436. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Handunnetthi, L.; Giovannoni, G.; Ramagopalan, S.V. An Updated Meta-Analysis of Risk of Multiple Sclerosis following Infectious Mononucleosis. PLoS ONE 2010, 5, e12496. [Google Scholar] [CrossRef] [PubMed]
- Hjalgrim, H.; Smedby, K.E.; Rostgaard, K.; Molin, D.; Hamilton-Dutoit, S.; Chang, E.T.; Ralfkiaer, E.; Sundstrom, C.; Adami, H.-O.; Glimelius, B.; et al. Infectious mononucleosis, childhood social environment, and risk of Hodgkin lymphoma. Cancer Res. 2007, 67, 2382–2388. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef]
- Healy, J.A.; Dave, S.S. The Role of EBV in the Pathogenesis of Diffuse Large B Cell Lymphoma. Curr. Top. Microbiol. Immunol. 2015, 390, 315–337. [Google Scholar] [CrossRef]
- Damania, B.; Kenney, S.C.; Raab-Traub, N. Epstein-Barr virus: Biology and clinical disease. Cell 2022, 185, 3652–3670. [Google Scholar] [CrossRef]
- Vereide, D.T.; Seto, E.; Chiu, Y.F.; Hayes, M.; Tagawa, T.; Grundhoff, A.; Hammerschmidt, W.; Sugden, B. Epstein-Barr virus maintains lymphomas via its miRNAs. Oncogene 2014, 33, 1258–1264. [Google Scholar] [CrossRef]
- Vereide, D.T.; Sugden, B. Lymphomas differ in their dependence on Epstein-Barr virus. Blood 2011, 117, 1977–1985. [Google Scholar] [CrossRef]
- Old, L.J.; Boyse, E.A.; Oettgen, H.F.; Harven, E.D.; Geering, G.; Williamson, B.; Clifford, P. Precipitating antibody in human serum to an antigen present in cultured burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 1966, 56, 1699–1704. [Google Scholar] [CrossRef]
- Wu, L.; Li, C.; Pan, L. Nasopharyngeal carcinoma: A review of current updates. Exp. Ther. Med. 2018, 15, 3687–3692. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; zur Hausen, H.; Becker, V. EB viral genomes in epithelial nasopharyngeal carcinoma cells. Nat. New Biol. 1973, 244, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H.; Schulte-Holthausen, H.; Klein, G.; Henle, W.; Henle, G.; Clifford, P.; Santesson, L. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 1970, 228, 1056–1058. [Google Scholar] [CrossRef] [PubMed]
- Hutt-Fletcher, L.M. The Long and Complicated Relationship between Epstein-Barr Virus and Epithelial Cells. J. Virol. 2017, 91, e01677-16. [Google Scholar] [CrossRef]
- Greenspan, J.S.; Greenspan, D.; Lennette, E.T.; Abrams, D.I.; Conant, M.A.; Petersen, V.; Freese, U.K. Replication of Epstein-Barr virus within the epithelial cells of oral “hairy” leukoplakia, an AIDS-associated lesion. N. Engl. J. Med. 1985, 313, 1564–1571. [Google Scholar] [CrossRef]
- Lemon, S.M.; Hutt, L.M.; Shaw, J.E.; Li, J.L.; Pagano, J.S. Replication of EBV in epithelial cells during infectious mononucleosis. Nature 1977, 268, 268–270. [Google Scholar] [CrossRef]
- Sixbey, J.W.; Nedrud, J.G.; Raab-Traub, N.; Hanes, R.A.; Pagano, J.S. Epstein-Barr virus replication in oropharyngeal epithelial cells. N. Engl. J. Med. 1984, 310, 1225–1230. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Chen, Y.-P.; Chan, A.T.C.; Quynh-Thu, L.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef]
- Niedobitek, G.; Hansmann, M.L.; Herbst, H.; Young, L.S.; Dienemann, D.; Hartmann, C.A.; Finn, T.; Pitteroff, S.; Welt, A.; Anagnostopoulos, I. Epstein-Barr virus and carcinomas: Undifferentiated carcinomas but not squamous cell carcinomas of the nasopharynx are regularly associated with the virus. J. Pathol. 1991, 165, 17–24. [Google Scholar] [CrossRef]
- Iizasa, H.; Nanbo, A.; Nishikawa, J.; Jinushi, M.; Yoshiyama, H. Epstein-Barr Virus (EBV)-associated Gastric Carcinoma. Viruses 2012, 4, 3420–3439. [Google Scholar] [CrossRef]
- Burke, A.P.; Yen, T.S.; Shekitka, K.M.; Sobin, L.H. Lymphoepithelial carcinoma of the stomach with Epstein-Barr virus demonstrated by polymerase chain reaction. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 1990, 3, 377–380. [Google Scholar]
- Sheen, T.-S.; Tsai, C.-C.; Ko, J.-Y.; Chang, Y.-L.; Hsu, M.-M. Undifferentiated carcinoma of the major salivary glands. Cancer 1997, 80, 357–363. [Google Scholar] [CrossRef]
- Tsang, C.M.; Deng, W.; Yip, Y.L.; Zeng, M.-S.; Lo, K.W.; Tsao, S.W. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin. J. Cancer 2014, 33, 549–555. [Google Scholar] [CrossRef]
- Feederle, R.; Neuhierl, B.; Bannert, H.; Geletneky, K.; Shannon-Lowe, C.; Delecluse, H.-J. Epstein-Barr virus B95.8 produced in 293 cells shows marked tropism for differentiated primary epithelial cells and reveals interindividual variation in susceptibility to viral infection. Int. J. Cancer 2007, 121, 588–594. [Google Scholar] [CrossRef]
- Sengupta, S.; den Boon, J.A.; Chen, I.H.; Newton, M.A.; Dahl, D.B.; Chen, M.; Cheng, Y.-J.; Westra, W.H.; Chen, C.-J.; Hildesheim, A.; et al. Genome-wide expression profiling reveals EBV-associated inhibition of MHC class I expression in nasopharyngeal carcinoma. Cancer Res. 2006, 66, 7999–8006. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, S.; Sun, R.; Wu, T.; Qian, J. Understanding the interplay between host immunity and Epstein-Barr virus in NPC patients. Emerg. Microbes Infect. 2015, 4, 1–9. [Google Scholar] [CrossRef]
- Pathmanthan, R.; Prasad, U.; Sadler, R.; Flynn, K.; Raab-Traub, N. Clonal proliferations of cells infected with Epstein-Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N. Engl. J. Med. 1995, 333, 693–698. [Google Scholar] [CrossRef]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.Y.; Takada, K.; Lui, V.W.Y.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef]
- Kimura, H. EBV in T-/NK-Cell Tumorigenesis. Adv. Exp. Med. Biol. 2018, 1045, 459–475. [Google Scholar] [CrossRef]
- Jones, J.F.; Shurin, S.; Abramowsky, C.; Tubbs, R.R.; Sciotto, C.G.; Wahl, R.; Sands, J.; Gottman, D.; Katz, B.Z.; Sklar, J. T-cell lymphomas containing Epstein-Barr viral DNA in patients with chronic Epstein-Barr virus infections. N. Engl. J. Med. 1988, 318, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Montes-Mojarro, I.A.; Fend, F.; Quintanilla-Martinez, L. EBV and the Pathogenesis of NK/T Cell Lymphoma. Cancers 2021, 13, 1414. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Ono, Y. Isolation of Epstein-Barr virus-infected clones of the human T-cell line MT-2: Use of recombinant viruses with a positive selection marker. J. Virol. 1995, 69, 3900–3903. [Google Scholar] [CrossRef]
- Imai, S.; Sugiura, M.; Oikawa, O.; Koizumi, S.; Hirao, M.; Kimura, H.; Hayashibara, H.; Terai, N.; Tsutsumi, H.; Oda, T.; et al. Epstein-Barr virus (EBV)-carrying and -expressing T-cell lines established from severe chronic active EBV infection. Blood 1996, 87, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.L.K.; Kelleher, C.; Amankonah, T.D.; Streib, J.E.; Xu, J.W.; Jones, J.F.; Gelfand, E.W. Model of Epstein-Barr virus infection of human thymocytes: Expression of viral genome and impact on cellular receptor expression in the T-lymphoblastic cell line, HPB-ALL. Blood 1995, 85, 456–464. [Google Scholar] [CrossRef]
- Tsuchiyama, J.; Yoshino, T.; Mori, M.; Kondoh, E.; Oka, T.; Akagi, T.; Hiraki, A.; Nakayama, H.; Shibuya, A.; Ma, Y.; et al. Characterization of a novel human natural killer-cell line (NK-YS) established from natural killer cell lymphoma/leukemia associated with Epstein-Barr virus infection. Blood 1998, 92, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.M.; Vera-Lozada, G.; Segges, P.; Hassan, R.; Niedobitek, G. Revisiting the Tissue Microenvironment of Infectious Mononucleosis: Identification of EBV Infection in T Cells and Deep Characterization of Immune Profiles. Front. Immunol. 2019, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Jonigk, D.; Laenger, F.; Maegel, L.; Izykowski, N.; Rische, J.; Tiede, C.; Klein, C.; Maecker-Kolhoff, B.; Kreipe, H.; Hussein, K. Molecular and Clinicopathological Analysis of Epstein-Barr Virus-Associated Posttransplant Smooth Muscle Tumors. Am. J. Transplant. 2012, 12, 1908–1917. [Google Scholar] [CrossRef]
- Lee, E.S.; Locker, J.; Nalesnik, M.; Reyes, J.; Jaffe, R.; Alashari, M.; Nour, B.; Tzakis, A.; Dickman, P.S. The association of Epstein-Barr virus with smooth-muscle tumors occurring after organ transplantation. N. Engl. J. Med. 1995, 332, 19–25. [Google Scholar] [CrossRef]
- Magg, T.; Schober, T.; Walz, C.; Ley-Zaporozhan, J.; Facchetti, F.; Klein, C.; Hauck, F. Epstein-Barr Virus(+) Smooth Muscle Tumors as Manifestation of Primary Immunodeficiency Disorders. Front. Immunol. 2018, 9, 368. [Google Scholar] [CrossRef]
- McClain, K.L.; Leach, C.T.; Jenson, H.B.; Joshi, V.V.; Pollock, B.H.; Parmley, R.T.; Dicarlo, F.J.; Chadwick, E.G.; Murphy, S.B. Association of Epstein-Barr virus with leiomyosarcomas in young people with AIDS. N. Engl. J. Med. 1995, 332, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Purgina, B.; Rao, U.N.M.; Miettinen, M.; Pantanowitz, L. AIDS-Related EBV-Associated Smooth Muscle Tumors: A Review of 64 Published Cases. Pathol. Res. Int. 2011, 2011, 561548. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.-S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.-S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef] [PubMed]
- James, J.A.; Harley, J.B.; Scofield, R.H. Epstein-Barr virus and systemic lupus erythematosus. Curr. Opin. Rheumatol. 2006, 18, 462–467. [Google Scholar] [CrossRef]
- Maslinska, M. The role of Epstein-Barr virus infection in primary Sjogren’s syndrome. Curr. Opin. Rheumatol. 2019, 31, 475–483. [Google Scholar] [CrossRef]
- Poole, B.D.; Scofield, R.H.; Harley, J.B.; James, J.A. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 2006, 39, 63–70. [Google Scholar] [CrossRef]
- Tsubota, K.; Fujishima, H.; Toda, I.; Katagiri, S.; Kawashima, Y.; Saito, I. Increased levels of Epstein-Barr virus DNA in lacrimal glands of Sjogren’s syndrome patients. Acta Ophthalmol. Scand. 1995, 73, 425–430. [Google Scholar] [CrossRef]
- Mercer, J.; Lee, J.E.; Saphire, E.O.; Freeman, S.A. SnapShot: Enveloped Virus Entry. Cell 2020, 182, 786. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Szakonyi, G.; Klein, M.G.; Hannan, J.P.; Young, K.A.; Ma, R.Z.; Asokan, R.; Holers, V.M.; Chen, X.S. Structure of the Epstein-Barr virus major envelope glycoprotein. Nat. Struct. Mol. Biol. 2006, 13, 996–1001. [Google Scholar] [CrossRef]
- Prota, A.E.; Sage, D.R.; Stehle, T.; Fingeroth, J.D. The crystal structure of human CD21: Implications for Epstein-Barr virus and C3d binding. Proc. Natl. Acad. Sci. USA 2002, 99, 10641–10646. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, H.E.; Asokan, R.; Holers, V.M.; Perkins, S.J. The 15 SCR flexible extracellular domains of human complement receptor type 2 can mediate multiple ligand and antigen interactions. J. Mol. Biol. 2006, 362, 1132–1147. [Google Scholar] [CrossRef]
- Backovic, M.; Longnecker, R.; Jardetzky, T.S. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. USA 2009, 106, 2880–2885. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Wu, L.; Chai, Y.; Qi, J.; Tan, S.; Gao, G.F.; Song, H.; Yan, J. Molecular basis of EphA2 recognition by gHgL from gammaherpesviruses. Nat. Commun. 2020, 11, 5964. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, A.N.; Sorem, J.; Longnecker, R.; Jardetzky, T.S. Structure of Epstein-Barr Virus Glycoprotein 42 Suggests a Mechanism for Triggering Receptor-Activated Virus Entry. Structure 2009, 17, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- Sathiyamoorthy, K.; Jiang, J.; Mohl, B.S.; Chen, J.; Zhou, Z.H.; Longnecker, R.; Jardetzky, T.S. Inhibition of EBV-mediated membrane fusion by anti-gHgL antibodies. Proc. Natl. Acad. Sci. USA 2017, 114, E8703–E8710. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Kirschner, A.N.; Longnecker, R.; Jardetzky, T.S. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. USA 2010, 107, 22641–22646. [Google Scholar] [CrossRef]
- Seiradake, E.; Harlos, K.; Sutton, G.; Aricescu, A.R.; Jones, E.Y. An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat. Struct. Mol. Biol. 2010, 17, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Nagae, M.; Re, S.; Mihara, E.; Nogi, T.; Sugita, Y.; Takagi, J. Crystal structure of alpha 5 beta 1 integrin ectodomain: Atomic details of the fibronectin receptor. J. Cell Biol. 2012, 197, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L.M. Epstein-Barr virus entry. J. Virol. 2007, 81, 7825–7832. [Google Scholar] [CrossRef] [PubMed]
- Ogembo, J.G.; Kannan, L.; Ghiran, I.; Nicholson-Weller, A.; Finberg, R.W.; Tsokos, G.C.; Fingeroth, J.D. Human Complement Receptor Type 1/CD35 Is an Epstein-Barr Virus Receptor. Cell Rep. 2013, 3, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.; Weis, J.; Fearon, D.; Whang, Y.; Kieff, E. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 1987, 50, 203–213. [Google Scholar] [CrossRef]
- Erdei, A.; Kovacs, K.G.; Nagy-Balo, Z.; Lukacsi, S.; Macsik-Valent, B.; Kurucz, I.; Bajtay, Z. New aspects in the regulation of human B cell functions by complement receptors CR1, CR2, CR3 and CR4. Immunol. Lett. 2021, 237, 42–57. [Google Scholar] [CrossRef]
- Nemerow, G.R.; Mullen, J.J., 3rd; Dickson, P.W.; Cooper, N.R. Soluble recombinant CR2 (CD21) inhibits Epstein-Barr virus infection. J. Virol. 1990, 64, 1348–1352. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.; Whang, Y.; Sample, J.; Sears, A.; Kieff, E. Soluble gp350/220 and deletion mutant glycoproteins block Epstein-Barr virus adsorption to lymphocytes. J. Virol. 1988, 62, 4452–4464. [Google Scholar] [CrossRef]
- Janz, A.; Oezel, M.; Kurzeder, C.; Mautner, J.; Pich, D.; Kost, M.; Hammerschmidt, W.; Delecluse, H.J. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 2000, 74, 10142–10152. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, B.; Li, Y.; Zhu, W.; Akihisa, T.; Li, W.; Kikuchi, T.; Liu, W.; Feng, F.; Zhang, J. Prophylactic and Therapeutic EBV Vaccines: Major Scientific Obstacles, Historical Progress, and Future Direction. Vaccines 2021, 9, 1290. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr Virus: An Important Vaccine Target for Cancer Prevention. Sci. Transl. Med. 2011, 3, 107fs7. [Google Scholar] [CrossRef]
- Kanekiyo, M.; Bu, W.; Joyce, M.G.; Meng, G.; Whittle, J.R.R.; Baxa, U.; Yamamoto, T.; Narpala, S.; Todd, J.-P.; Rao, S.S.; et al. Rational Design of an Epstein-Barr Virus Vaccine Targeting the Receptor-Binding Site. Cell 2015, 162, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.J.; Lazarowitz, S.G.; Hayward, S.D. Monoclonal antibody against a 250,000-dalton glycoprotein of Epstein-Barr virus identifies a membrane antigen and a neutralizing antigen. Proc. Natl. Acad. Sci. USA 1980, 77, 2979–2983. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Heston, L.; Hoffman, G. Neutralization of lymphocyte immortalization by different strains of Epstein-Barr virus with a murine monoclonal antibody. Infect. Immun. 1982, 37, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.E.; Hu, J.; Alfieri, C. Construction and Characterization of a Humanized Anti-Epstein-Barr Virus gp350 Antibody with Neutralizing Activity in Cell Culture. Cancers 2018, 10, 112. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Geilinger, K. Monoclonal antibodies against the major glycoprotein (gp350/220) of Epstein-Barr virus neutralize infectivity. Proc. Natl. Acad. Sci. USA 1980, 77, 5307–5311. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M.; Hoppenbrouwers, K.; Vandermeulen, C.; Moutschen, M.; Leonard, P.; Moreels, A.; Haumont, M.; Bollen, A.; Smets, F.; Denis, M. Recombinant gp350 vaccine for infectious mononucleosis: A phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J. Infect. Dis. 2007, 196, 1749–1753. [Google Scholar] [CrossRef]
- Java, A.; Liszewski, M.K.; Hourcade, D.E.; Zhang, F.; Atkinson, J.P. Role of complement receptor 1 (CR1; CD35) on epithelial cells: A model for understanding complement-mediated damage in the kidney. Mol. Immunol. 2015, 67, 584–595. [Google Scholar] [CrossRef]
- Jiang, R.; Gu, X.; Nathan, C.-A.; Hutt-Fletcher, L. Laser-capture microdissection of oropharyngeal epithelium indicates restriction of Epstein-Barr virus receptor/CD21 mRNA to tonsil epithelial cells. J. Oral Pathol. Med. 2008, 37, 626–633. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Palefsky, J.M.; Herrera, R.; Tugizov, S.M. Characterization of the Epstein-Barr virus glycoprotein BMRF-2. Virology 2007, 359, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Hussein, H.A.M.; Walker, L.R.; Abdel-Raouf, U.M.; Desouky, S.A.; Montasser, A.K.M.; Akula, S.M. Beyond RGD: Virus interactions with integrins. Arch. Virol. 2015, 160, 2669–2681. [Google Scholar] [CrossRef]
- Xiao, J.; Palefsky, J.M.; Herrera, R.; Berline, J.; Tugizov, S.M. The Epstein-Barr virus BMRF-2 protein facilitates virus attachment to oral epithelial cells. Virology 2008, 370, 430–442. [Google Scholar] [CrossRef]
- Xiao, J.; Palefsky, J.M.; Herrera, R.; Berline, J.; Tugizov, S.M. EBV BMRF-2 facilitates cell-to-cell spread of virus within polarized oral epithelial cells. Virology 2009, 388, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.; Hutt-Fletcher, L.M. The BDLF2 protein of Epstein-Barr virus is a type II glycosylated envelope protein whose processing is dependent on coexpression with the BMRF2 protein. Virology 2009, 383, 162–167. [Google Scholar] [CrossRef]
- Chesnokova, L.S.; Hutt-Fletcher, L.M. Fusion of Epstein-Barr Virus with Epithelial Cells Can Be Triggered by alpha v beta 5 in Addition to alpha v beta 6 and alpha v beta 8, and Integrin Binding Triggers a Conformational Change in Glycoproteins gHgL. J. Virol. 2011, 85, 13214–13223. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt-Fletcher, L.M. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alpha v beta 6 or alpha v beta 8. Proc. Natl. Acad. Sci. USA 2009, 106, 20464–20469. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; White, B.E.P.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Spriggs, M.K.; Kovats, S.; Turk, S.M.; Comeau, M.R.; Nepom, B.; Hutt-Fletcher, L.M. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 1997, 71, 4657–4662. [Google Scholar] [CrossRef]
- Haan, K.M.; Kwok, W.W.; Longnecker, R.; Speck, P. Epstein-Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor. J. Virol. 2000, 74, 2451–2454. [Google Scholar] [CrossRef] [PubMed]
- Borza, C.M.; Hutt-Fletcher, L.M. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 2002, 8, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hutt-Fletcher, L.M. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J. Virol. 1998, 72, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Sathiyamoorthy, K.; Jiang, J.; Hu, Y.X.; Rowe, C.L.; Mohl, B.S.; Chen, J.; Jiang, W.; Mellins, E.D.; Longnecker, R.; Zhou, Z.H.; et al. Assembly and Architecture of the EBV B Cell Entry Triggering Complex. PLoS Pathog. 2014, 10, e1004309. [Google Scholar] [CrossRef] [PubMed]
- Borza, C.M.; Morgan, A.J.; Turk, S.M.; Hutt-Fletcher, L.M. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol. 2004, 78, 5007–5014. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Yamauchi, Y.; Teesalu, T. A widespread viral entry mechanism: The C-end Rule motif-neuropilin receptor interaction. Proc. Natl. Acad. Sci. USA 2021, 118, e2112457118. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.-B.; Braun, G.B.; Friman, T.; Aza-Blanc, P.; Ruidiaz, M.E.; Sugahara, K.N.; Teesalu, T.; Ruoslahti, E. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat. Commun. 2014, 5, 4904. [Google Scholar] [CrossRef]
- Wang, H.-B.; Zhang, H.; Zhang, J.-P.; Li, Y.; Zhao, B.; Feng, G.-K.; Du, Y.; Xiong, D.; Zhong, Q.; Liu, W.-L.; et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015, 6, 6240. [Google Scholar] [CrossRef]
- Tan, L.; Yuan, X.; Liu, Y.; Cai, X.; Guo, S.; Wang, A. Non-muscle Myosin II: Role in Microbial Infection and Its Potential as a Therapeutic Target. Front. Microbiol. 2019, 10, 401. [Google Scholar] [CrossRef]
- Xiong, D.; Du, Y.; Wang, H.-B.; Zhao, B.; Zhang, H.; Li, Y.; Hu, L.-J.; Cao, J.-Y.; Zhong, Q.; Liu, W.-L.; et al. Nonmuscle myosin heavy chain IIA mediates Epstein-Barr virus infection of nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2015, 112, 11036–11041. [Google Scholar] [CrossRef]
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EphA2 in cancer. J. Hematol. Oncol. 2020, 13, 114. [Google Scholar] [CrossRef]
- Du Toit, A. Cellular microbiology: Many pathogens, one host receptor. Nat. Rev. Microbiol. 2018, 16, 64. [Google Scholar] [CrossRef]
- Hahn, A.S.; Kaufmann, J.K.; Wies, E.; Naschberger, E.; Panteleev-Ivlev, J.; Schmidt, K.; Holzer, A.; Schmidt, M.; Chen, J.; Koenig, S.; et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 2012, 18, 961–966. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Wang, H.-B.; Zhang, A.; Chen, M.-L.; Fang, Z.-X.; Dong, X.-D.; Li, S.-B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol. 2018, 3, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.T.; Dutch, R.E. Viral Membrane Fusion and the Transmembrane Domain. Viruses 2020, 12, 693. [Google Scholar] [CrossRef] [PubMed]
- Baquero, E.; Albertini, A.A.V.; Gaudin, Y. Recent mechanistic and structural insights on class III viral fusion glycoproteins. Curr. Opin. Struct. Biol. 2015, 33, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Molesworth, S.J.; Lake, C.M.; Borza, C.M.; Turk, S.M.; Hutt-Fletcher, L.M. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 2000, 74, 6324–6332. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Viral Protein | Receptor/Binding Factor | Target Cell | Function | Structure |
|---|---|---|---|---|
| Membrane protein | ||||
| BMRF2 | Integrins | Epithelial cell | Viral attachment | None available complex structure |
| Glycoproteins | ||||
| gp350 | CD21 | B lymphocyte | Viral attachment | gp350 ecto-domain (PDB:2H6O) [95] CD21 SCR1-SCR2 (PDB: 1LY2) [96] CD21 (PDB: 2GSX) [97] None available complex structure |
| CD35 | B lymphocyte | Viral attachment | None available complex structure | |
| gB | NRP1 | Epithelial cell | Viral binding and promote membrane fusion | Post-fusion gB extracellular domain (PDB: 3FVC) [98] None available pre-gB structure None available complex structure |
| EphA2 | Epithelial cell | Viral binding and promote membrane fusion | None available complex structure | |
| BMRF2 | Integrins | Epithelial cell | Viral attachment | None available complex structure |
| gHgL | Integrins | Epithelial cell | Viral attachment | None available complex structure |
| NMHC-IIA | Epithelial cell | Viral binding and promote membrane fusion | None available complex structure | |
| EphA2 | Epithelial cell | Viral binding and promote membrane fusion | gHgL-EphA2(Ligand binding domain) (PDB: 7CZE) [99] | |
| gHgL-gp42 | HLAII | B lymphocyte | Viral binding and promote membrane fusion | gp42 (PDB: 3FD4) [100] gp42-HLA-DR1 (PDB: 1KG0) [101] gHgL-CL40-gp42_N-domain (PDB: 5W0K) [102] gHgL-gp42-E1D1 (PDB: 5T1D) [17] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bu, G.-L.; Xie, C.; Kang, Y.-F.; Zeng, M.-S.; Sun, C. How EBV Infects: The Tropism and Underlying Molecular Mechanism for Viral Infection. Viruses 2022, 14, 2372. https://doi.org/10.3390/v14112372
Bu G-L, Xie C, Kang Y-F, Zeng M-S, Sun C. How EBV Infects: The Tropism and Underlying Molecular Mechanism for Viral Infection. Viruses. 2022; 14(11):2372. https://doi.org/10.3390/v14112372
Chicago/Turabian StyleBu, Guo-Long, Chu Xie, Yin-Feng Kang, Mu-Sheng Zeng, and Cong Sun. 2022. "How EBV Infects: The Tropism and Underlying Molecular Mechanism for Viral Infection" Viruses 14, no. 11: 2372. https://doi.org/10.3390/v14112372
APA StyleBu, G.-L., Xie, C., Kang, Y.-F., Zeng, M.-S., & Sun, C. (2022). How EBV Infects: The Tropism and Underlying Molecular Mechanism for Viral Infection. Viruses, 14(11), 2372. https://doi.org/10.3390/v14112372