
G-Quadruplex DNA and Other Non-Canonical B-Form DNA Motifs Influence Productive and Latent HIV-1 Integration and Reactivation Potential


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
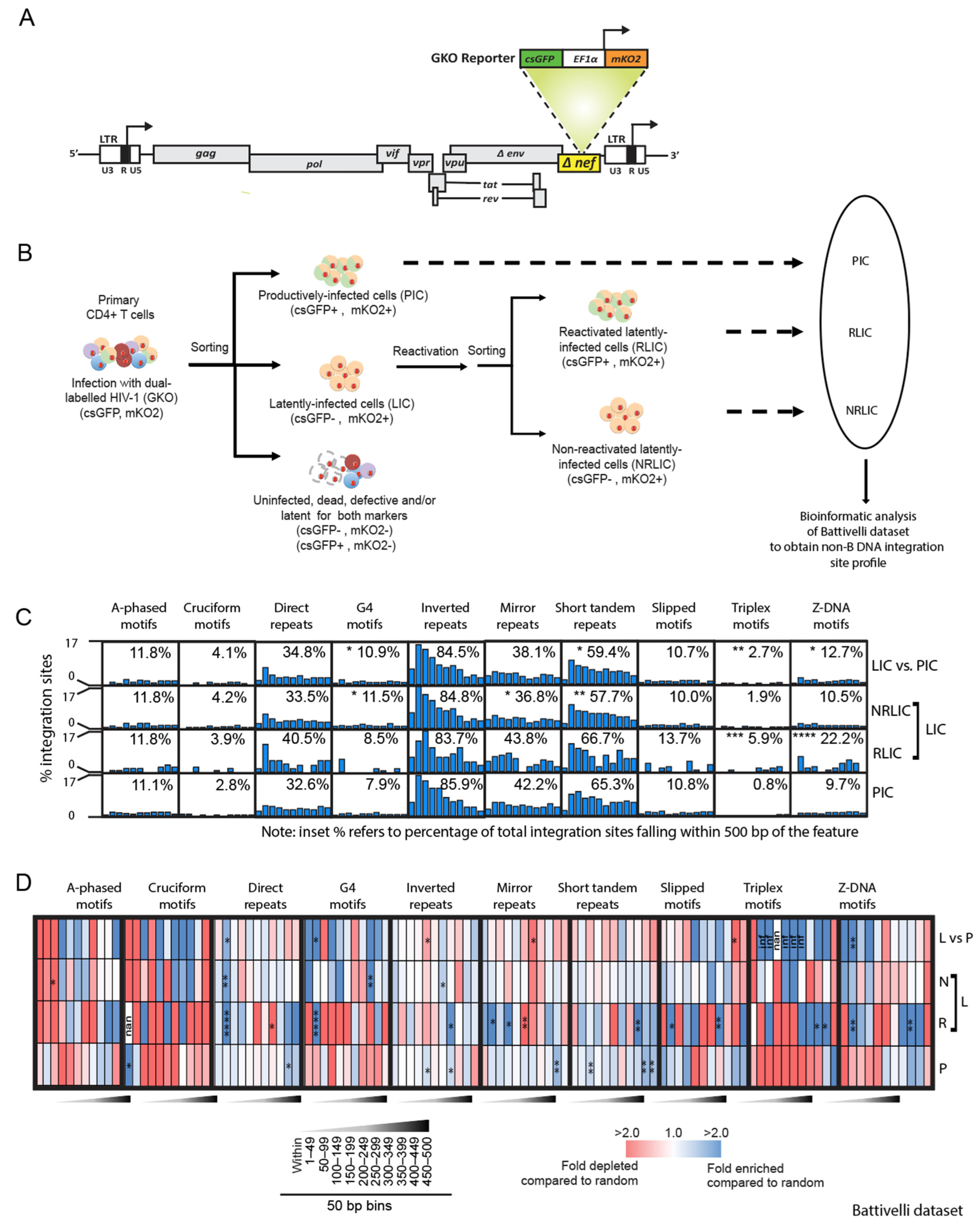
2.1. Cell Propagation, Virus Production and Infection
2.2. Drug Treatment and Infection for Integration Site Analysis
2.3. HIV Integration Site Library and Computational Analysis
2.4. MTT Assay
2.5. Confocal Immunofluorescence Microscopy
2.6. Flow Cytometry
2.7. Datasets
2.8. Statistical Analyses
3. Results
3.1. Reactivation Potential of HIV-1 Proviruses Correlates with Integration Site Placement near Non-B DNA Motifs
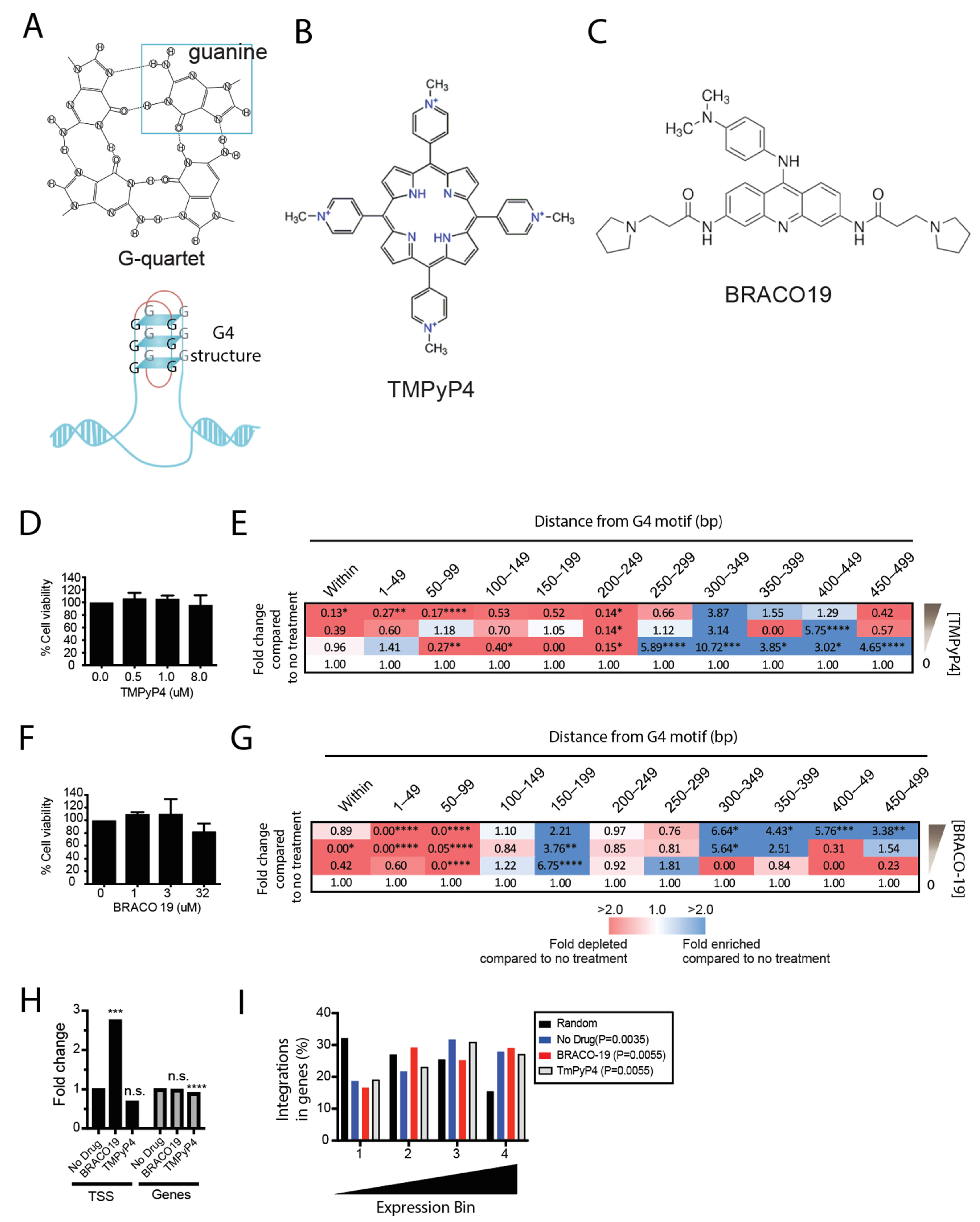
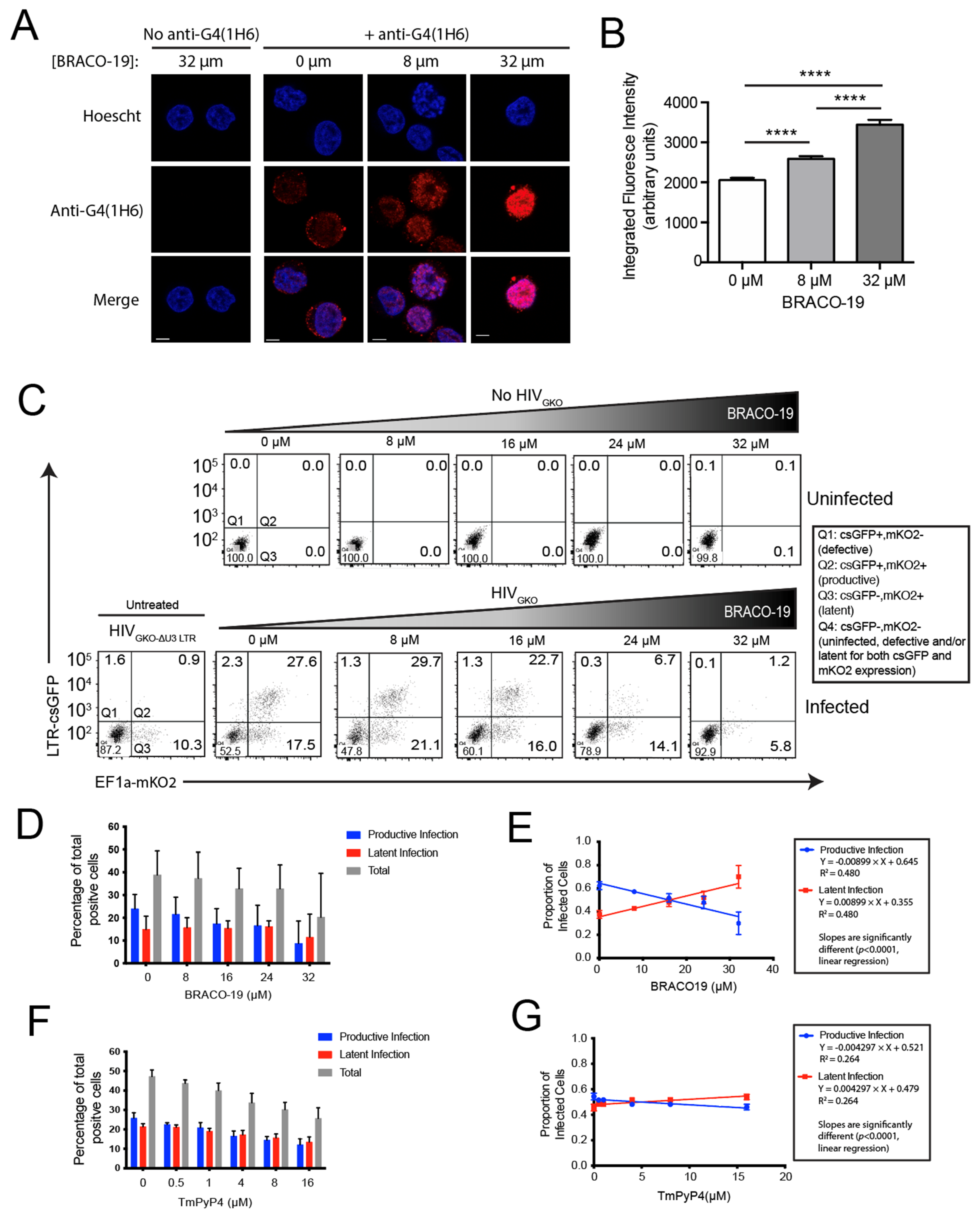
3.2. Pretreatment of Cells with G4 Ligands Alters HIV-1 Integration Targeting of G4 DNA
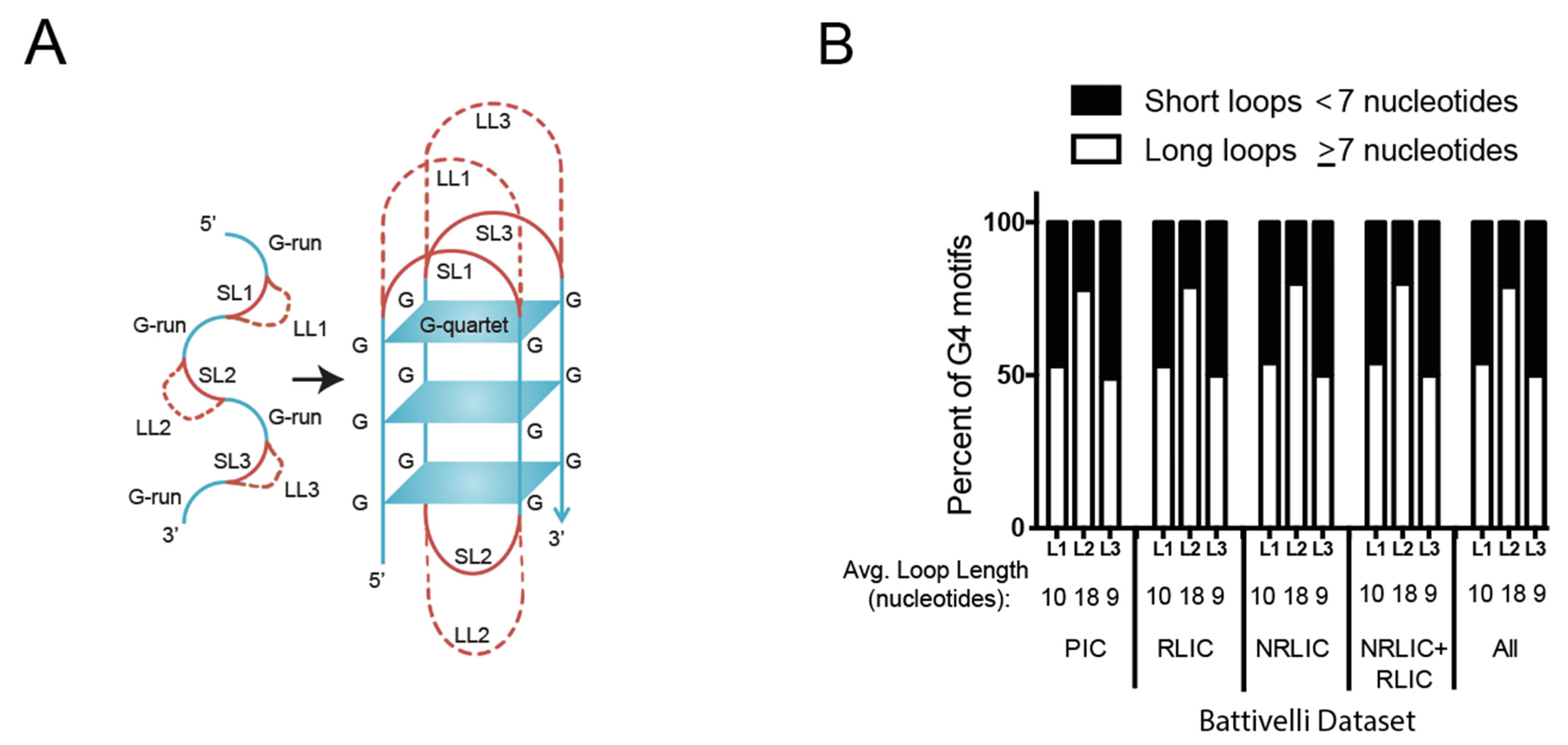
3.3. HIV-1 Favors G4 Structures with Long-Loops for Integration
3.4. Stabilization of G4 Structures In Vitro Increases the Proportion of Latently Infected Cells
3.5. CPSF6 and LEDGF/p75 Influence Integration near Non-B DNA Motifs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arts, E.J.; Hazuda, D.J. HIV-1 Antiretroviral Drug Therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In Vivo Fate of HIV-1-Infected T Cells: Quantitative Analysis of the Transition to Stable Latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early Establishment of a Pool of Latently Infected, Resting CD4(+) T Cells during Primary HIV-1 Infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T Cell Dynamics after Interruption of Highly Active Antiretroviral Therapy (HAART) in Patients with a History of Sustained Viral Suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [Green Version]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of Vorinostat Disrupts HIV-1 Latency in Patients on Antiretroviral Therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [Green Version]
- Deeks, S.G. HIV: Shock and Kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent Infection of CD4+ T Cells Provides a Mechanism for Lifelong Persistence of HIV-1, Even in Patients on Effective Combination Therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-Term Follow-up Studies Confirm the Stability of the Latent Reservoir for HIV-1 in Resting CD4+ T Cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Archin, N.M.; Vaidya, N.K.; Kuruc, J.D.; Liberty, A.L.; Wiegand, A.; Kearney, M.F.; Cohen, M.S.; Coffin, J.M.; Bosch, R.J.; Gay, C.L.; et al. Immediate Antiviral Therapy Appears to Restrict Resting CD4+ Cell HIV-1 Infection without Accelerating the Decay of Latent Infection. Proc. Natl. Acad. Sci. USA 2012, 109, 9523–9528. [Google Scholar] [CrossRef] [Green Version]
- Rong, L.; Perelson, A.S. Modeling Latently Infected Cell Activation: Viral and Latent Reservoir Persistence, and Viral Blips in HIV-Infected Patients on Potent Therapy. PLoS Comput. Biol. 2009, 5, e1000533. [Google Scholar] [CrossRef]
- Palmer, S.; Wiegand, A.P.; Maldarelli, F.; Bazmi, H.; Mican, J.M.; Polis, M.; Dewar, R.L.; Planta, A.; Liu, S.; Metcalf, J.A.; et al. New Real-Time Reverse Transcriptase-Initiated PCR Assay with Single-Copy Sensitivity for Human Immunodeficiency Virus Type 1 RNA in Plasma. J. Clin. Microbiol. 2003, 41, 4531–4536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruelas, D.S.; Greene, W.C. An Integrated Overview of HIV-1 Latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV Latency: The Road to an HIV Cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamer, D.H. Can HIV Be Cured? Mechanisms of HIV Persistence and Strategies to Combat It. Curr. HIV Res. 2004, 2, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geeraert, L.; Kraus, G.; Pomerantz, R.J. Hide-and-Seek: The Challenge of Viral Persistence in HIV-1 Infection. Annu. Rev. Med. 2008, 59, 487–501. [Google Scholar] [CrossRef]
- Savarino, A.; Mai, A.; Norelli, S.; El Daker, S.; Valente, S.; Rotili, D.; Altucci, L.; Palamara, A.T.; Garaci, E. “Shock and Kill” Effects of Class I-Selective Histone Deacetylase Inhibitors in Combination with the Glutathione Synthesis Inhibitor Buthionine Sulfoximine in Cell Line Models for HIV-1 Quiescence. Retrovirology 2009, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, T.A.; Lewin, S.R. Shocking HIV out of Hiding: Where Are We with Clinical Trials of Latency Reversing Agents? Curr. Opin. HIV AIDS 2016, 11, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; Svensson, J.P.; Tojal Da Silva, I.; Cohn, L.B.; Gramatica, A.; Deeks, S.; Greene, W.C.; Pillai, S.K.; et al. Distinct Chromatin Functional States Correlate with HIV Latency Reactivation in Infected Primary CD4+ T Cells. Elife 2018, 7, e34655. [Google Scholar] [CrossRef]
- Jordan, A.; Defechereux, P.; Verdin, E. The Site of HIV-1 Integration in the Human Genome Determines Basal Transcriptional Activity and Response to Tat Transactivation. EMBO J. 2001, 20, 1726–1738. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-C.; Martinez, J.P.; Zorita, E.; Meyerhans, A.; Filion, G.J. Position Effects Influence HIV Latency Reversal. Nat. Struct. Mol. Biol. 2017, 24, 47–54. [Google Scholar] [CrossRef]
- Sherrill-Mix, S.; Lewinski, M.K.; Famiglietti, M.; Bosque, A.; Malani, N.; Ocwieja, K.E.; Berry, C.C.; Looney, D.; Shan, L.; Agosto, L.M.; et al. HIV Latency and Integration Site Placement in Five Cell-Based Models. Retrovirology 2013, 10, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahabieh, M.S.; Ooms, M.; Brumme, C.; Taylor, J.; Harrigan, P.R.; Simon, V.; Sadowski, I. Direct Non-Productive HIV-1 Infection in a T-Cell Line Is Driven by Cellular Activation State and NFκB. Retrovirology 2014, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. Specific HIV Integration Sites Are Linked to Clonal Expansion and Persistence of Infected Cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetti, F.R.; Sobolewski, M.D.; Fyne, E.; Shao, W.; Spindler, J.; Hattori, J.; Anderson, E.M.; Watters, S.A.; Hill, S.; Wu, X.; et al. Clonally Expanded CD4 + T Cells Can Produce Infectious HIV-1 in Vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1883–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef] [Green Version]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective Proviruses Rapidly Accumulate during Acute HIV-1 Infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A Role for LEDGF/P75 in Targeting HIV DNA Integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef]
- Shun, M.-C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/P75 Functions Downstream from Preintegration Complex Formation to Effect Gene-Specific HIV-1 Integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/P75 in Lentiviral Infectivity and Integration Targeting. PLoS ONE 2007, 2, e1340. [Google Scholar] [CrossRef] [Green Version]
- Vranckx, L.S.; Demeulemeester, J.; Saleh, S.; Boll, A.; Vansant, G.; Schrijvers, R.; Weydert, C.; Battivelli, E.; Verdin, E.; Cereseto, A.; et al. LEDGIN-Mediated Inhibition of Integrase–LEDGF/P75 Interaction Reduces Reactivation of Residual Latent HIV. EBioMedicine 2016, 8, 248–264. [Google Scholar] [CrossRef]
- Singh, P.K.; Plumb, M.R.; Ferris, A.L.; Iben, J.R.; Wu, X.; Fadel, H.J.; Luke, B.T.; Esnault, C.; Poeschla, E.M.; Hughes, S.H.; et al. LEDGF/P75 Interacts with MRNA Splicing Factors and Targets HIV-1 Integration to Highly Spliced Genes. Genes Dev. 2015, 29, 2287–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowd, G.A.; Serrao, E.; Wang, H.; Wang, W.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. A Critical Role for Alternative Polyadenylation Factor CPSF6 in Targeting HIV-1 Integration to Transcriptionally Active Chromatin. Proc. Natl. Acad. Sci. USA 2016, 113, E1054–E1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Singh, P.K.; Sowd, G.A.; Bedwell, G.J.; Jang, S.; Achuthan, V.; Oleru, A.V.; Wong, D.; Fadel, H.J.; Lee, K.; et al. CPSF6-Dependent Targeting of Speckle-Associated Domains Distinguishes Primate from Nonprimate Lentiviral Integration. mBio 2020, 11, e02254-20. [Google Scholar] [CrossRef] [PubMed]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.-C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV Reporducibly Establishes a Latent Infection after Acute Infection of T Cells Ni Vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Einkauf, K.B.; Lee, G.Q.; Gao, C.; Sharaf, R.; Sun, X.; Hua, S.; Chen, S.M.Y.M.; Jiang, C.; Lian, X.; Chowdhury, F.Z.; et al. Intact HIV-1 Proviruses Accumulate at Distinct Chromosomal Positions during Prolonged Antiretroviral Therapy. J. Clin. Investig. 2019, 129, 988–998. [Google Scholar] [CrossRef] [Green Version]
- McAllister, R.G.; Liu, J.; Woods, M.W.; Tom, S.K.; Rupar, C.A.; Barr, S.D. Lentivector Integration Sites in Ependymal Cells from a Model of Metachromatic Leukodystrophy: Non-B DNA as a New Factor Influencing Integration. Mol. Nucleic Acids 2014, 3, e187. [Google Scholar] [CrossRef]
- Bacolla, A.; Wells, R.D. Non-B DNA Conformations, Genomic Rearrangements, and Human Disease. J. Biol. Chem. 2004, 279, 47411–47414. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Majima, T. Conformational Changes of Non-BDNA. Chem. Soc. Rev. 2011, 40, 5893–5909. [Google Scholar] [CrossRef]
- Wahls, W.P.; Wallace, L.J.; Moore, P.D. The Z-DNA Motif d(TG)30 Promotes Reception of Information during Gene Conversion Events While Stimulating Homologous Recombination in Human Cells in Culture. Mol. Cell. Biol. 1990, 10, 785–793. [Google Scholar] [PubMed] [Green Version]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct Evidence for a G-Quadruplex in a Promoter Region and Its Targeting with a Small Molecule to Repress c-MYC Transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Yadav, V.K.; Basundra, R.; Kumar, A.; Chowdhury, S. Evidence of Genome-Wide G4 DNA-Mediated Gene Expression in Human Cancer Cells. Nucleic Acids Res 2009, 37, 4194–4204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waga, S.; Mizuno, S.; Yoshida, M. Chromosomal Protein HMG1 Removes the Transcriptional Block Caused by the Cruciform in Supercoiled DNA. J. Biol. Chem. 1990, 265, 19424–19428. [Google Scholar] [CrossRef]
- Waga, S.; Mizuno, S.; Yoshida, M. Nonhistone Protein HMG1 Removes the Transcriptional Block Caused by Left-Handed Z-Form Segment in a Supercoiled DNA. Biochem. Biophys. Res. Commun. 1988, 153, 334–339. [Google Scholar] [CrossRef]
- Jain, A.; Magistri, M.; Napoli, S.; Carbone, G.M.; Catapano, C.V. Mechanisms of Triplex DNA-Mediated Inhibition of Transcription Initiation in Cells. Biochimie 2010, 92, 317–320. [Google Scholar] [CrossRef]
- Maher, L.J.; Dervan, P.B.; Wold, B. Analysis of Promoter-Specific Repression by Triple-Helical DNA Complexes in a Eukaryotic Cell-Free Transcription System. Biochemistry 1992, 31, 70–81. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA Secondary Structures: Stability and Function of G-Quadruplex Structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [Green Version]
- Brázda, V.; Laister, R.C.; Jagelská, E.B.; Arrowsmith, C. Cruciform Structures Are a Common DNA Feature Important for Regulating Biological Processes. BMC Mol. Biol. 2011, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Delic, J.; Onclercq, R.; Moisan-Coppey, M. Inhibition and Enhancement of Eukaryotic Gene Expression by Potential Non-B DNA Sequences. Biochem. Biophys. Res. Commun. 1991, 180, 1273–1283. [Google Scholar] [CrossRef]
- Tornaletti, S.; Park-Snyder, S.; Hanawalt, P.C. G4-Forming Sequences in the Non-Transcribed DNA Strand Pose Blocks to T7 RNA Polymerase and Mammalian RNA Polymerase II. J. Biol. Chem. 2008, 283, 12756–12762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belotserkovskii, B.P.; De Silva, E.; Tornaletti, S.; Wang, G.; Vasquez, K.M.; Hanawalt, P.C. A Triplex-Forming Sequence from the Human c-MYC Promoter Interferes with DNA Transcription. J. Biol. Chem. 2007, 282, 32433–32441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, S.D.; Ciuffi, A.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. HIV Integration Site Selection: Targeting in Macrophages and the Effects of Different Routes of Viral Entry. Mol. Ther. 2006, 14, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Perrone, R.; Butovskaya, E.; Daelemans, D.; Palù, G.; Pannecouque, C.; Richter, S.N. Anti-HIV-1 Activity of the G-Quadruplex Ligand BRACO-19. J. Antimicrob. Chemother. 2014, 69, 3248–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofer, N.; Weisman-Shomer, P.; Shklover, J.; Fry, M. The Quadruplex r(CGG)n Destabilizing Cationic Porphyrin TMPyP4 Cooperates with HnRNPs to Increase the Translation Efficiency of Fragile X Premutation MRNA. Nucleic Acids Res. 2009, 37, 2712–2722. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.J.; Wingate, K.L.; Silwal, J.; Leeper, T.C.; Basu, S. The Porphyrin TmPyP4 Unfolds the Extremely Stable G-Quadruplex in MT3-MMP MRNA and Alleviates Its Repressive Effect to Enhance Translation in Eukaryotic Cells. Nucleic Acids Res. 2012, 40, 4137–4145. [Google Scholar] [CrossRef]
- Ciuffi, A.; Barr, S.D. Identification of HIV Integration Sites in Infected Host Genomic DNA. Methods 2011, 53, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Schroder, A.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F.D. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef] [Green Version]
- Cer, R.Z.; Donohue, D.E.; Mudunuri, U.S.; Temiz, N.A.; Loss, M.A.; Starner, N.J.; Halusa, G.N.; Volfovsky, N.; Yi, M.; Luke, B.T.; et al. Non-B DB v2.0: A Database of Predicted Non-B DNA-Forming Motifs and Its Associated Tools. Nucleic Acids Res. 2013, 41, D94–D100. [Google Scholar] [CrossRef]
- Kazemier, H.G.; Paeschke, K.; Lansdorp, P.M. Guanine Quadruplex Monoclonal Antibody 1H6 Cross-Reacts with Restrained Thymidine-Rich Single Stranded DNA. Nucleic Acids Res. 2017, 45, 5913–5919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, A.; Wu, Y.; Huang, Y.C.; Chavez, E.A.; Platt, J.; Johnson, F.B.; Brosh, R.M.; Sen, D.; Lansdorp, P.M. Detection of G-Quadruplex DNA in Mammalian Cells. Nucleic Acids Res. 2014, 42, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Du, Z.; Li, N. Extensive Selection for the Enrichment of G4 DNA Motifs in Transcriptional Regulatory Regions of Warm Blooded Animals. FEBS Lett. 2007, 581, 1951–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Zhao, Y.; Li, N. Genome-Wide Analysis Reveals Regulatory Role of G4 DNA in Gene Transcription. Genome Res. 2008, 18, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Kouzine, F.; Wojtowicz, D.; Baranello, L.; Yamane, A.; Nelson, S.; Resch, W.; Kieffer-Kwon, K.-R.; Benham, C.J.; Casellas, R.; Przytycka, T.M.; et al. Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome. Cell Syst. 2017, 4, 344–356.e7. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative Visualization of DNA G-Quadruplex Structures in Human Cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Perrone, R.; Nadai, M.; Poe, J.A.; Frasson, I.; Palumbo, M.; Palù, G.; Smithgall, T.E.; Richter, S.N. Formation of a Unique Cluster of G-Quadruplex Structures in the HIV-1 Nef Coding Region: Implications for Antiviral Activity. PLoS Med. 2013, 8, e73121. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L. Structure, Location and Interactions of G-Quadruplexes. FEBS J. 2010, 277, 3452–3458. [Google Scholar] [CrossRef]
- Weisman-Shomer, P.; Cohen, E.; Hershco, I.; Khateb, S.; Wolfovitz-Barchad, O.; Hurley, L.H.; Fry, M. The Cationic Porphyrin TMPyP4 Destabilizes the Tetraplex Form of the Fragile X Syndrome Expanded Sequence d(CGG)n. Nucleic Acids Res. 2003, 31, 3963–3970. [Google Scholar] [CrossRef]
- Han, H.; Langley, D.R.; Rangan, A.; Hurley, L.H. Selective Interactions of Cationic Porphyrins with G-Quadruplex Structures. J. Am. Chem. Soc. 2001, 123, 8902–8913. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-H.; Chen, S.-B.; Wang, B.; Ou, T.-M.; Gu, L.-Q.; Tan, J.-H.; Huang, Z.-S. Specific Targeting of Telomeric Multimeric G-Quadruplexes by a New Triaryl-Substituted Imidazole. Nucleic Acids Res. 2017, 45, 1606–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grand, C.L.; Han, H.; Muñoz, R.M.; Weitman, S.; Von Hoff, D.D.; Hurley, L.H.; Bearss, D.J. The Cationic Porphyrin TMPyP4 Down-Regulates c-MYC and Human Telomerase Reverse Transcriptase Expression and Inhibits Tumor Growth in Vivo. Mol. Cancer 2002, 1, 565–573. [Google Scholar]
- White, E.W.; Tanious, F.; Ismail, M.A.; Reszka, A.P.; Neidle, S.; Boykin, D.W.; Wilson, W.D. Structure-Specific Recognition of Quadruplex DNA by Organic Cations: Influence of Shape, Substituents and Charge. Biophys. Chem. 2007, 126, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Read, M.; Harrison, R.J.; Romagnoli, B.; Tanious, F.A.; Gowan, S.H.; Reszka, A.P.; Wilson, W.D.; Kelland, L.R.; Neidle, S. Structure-Based Design of Selective and Potent G Quadruplex-Mediated Telomerase Inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 4844–4849. [Google Scholar] [CrossRef] [Green Version]
- Burger, A.M.; Dai, F.; Schultes, C.M.; Reszka, A.P.; Moore, M.J.; Double, J.A.; Neidle, S. The G-Quadruplex-Interactive Molecule BRACO-19 Inhibits Tumor Growth, Consistent with Telomere Targeting and Interference with Telomerase Function. Cancer Res. 2005, 65, 1489–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tippana, R.; Hwang, H.; Opresko, P.L.; Bohr, V.A.; Myong, S. Single-Molecule Imaging Reveals a Common Mechanism Shared by G-Quadruplex-Resolving Helicases. Proc. Natl. Acad. Sci. USA 2016, 113, 8448–8453. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L.; Balasubramanian, S. G-Quadruplexes in Promoters throughout the Human Genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef] [Green Version]
- Maizels, N.; Gray, L.T.; Gellert, M.; Lipsett, M.N.; Davies, D.R.; Sen, D.; Gilbert, W.; Kim, J.; Cheong, C.; Moore, P.B.; et al. The G4 Genome. PLoS Genet. 2013, 9, e1003468. [Google Scholar] [CrossRef] [Green Version]
- Eddy, J.; Vallur, A.C.; Varma, S.; Liu, H.; Reinhold, W.C.; Pommier, Y.; Maizels, N. G4 Motifs Correlate with Promoter-Proximal Transcriptional Pausing in Human Genes. Nucleic Acids Res. 2011, 39, 4975–4983. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Zhao, Y.; Li, N. Genome-Wide Colonization of Gene Regulatory Elements by G4 DNA Motifs. Nucleic Acids Res. 2009, 37, 6784–6798. [Google Scholar] [CrossRef] [PubMed]
- Eddy, J.; Maizels, N. Selection for the G4 DNA Motif at the 5′ End of Human Genes. Mol. Carcinog. 2009, 48, 319–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A Web-Based Server for Predicting G-Quadruplexes in Nucleotide Sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef] [PubMed]
- Scaria, V.; Hariharan, M.; Arora, A.; Maiti, S. Quadfinder: Server for Identification and Analysis of Quadruplex-Forming Motifs in Nucleotide Sequences. Nucleic Acids Res. 2006, 34, W683–W685. [Google Scholar] [CrossRef] [Green Version]
- Bugaut, A.; Balasubramanian, S. A Sequence-Independent Study of the Influence of Short Loop Lengths on the Stability and Topology of Intramolecular DNA G-Quadruplexes. Biochemistry 2008, 47, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Frees, S.; Menendez, C.; Crum, M.; Bagga, P.S. QGRS-Conserve: A Computational Method for Discovering Evolutionarily Conserved G-Quadruplex Motifs. Hum. Genom. 2014, 8, 8. [Google Scholar] [CrossRef]
- Puig Lombardi, E.; Londoño-Vallejo, A. A Guide to Computational Methods for G-Quadruplex Prediction. Nucleic Acids Res. 2020, 48, 1–15. [Google Scholar] [CrossRef]
- Lago, S.; Tosoni, E.; Nadai, M.; Palumbo, M.; Richter, S.N. The Cellular Protein Nucleolin Preferentially Binds Long-Looped G-Quadruplex Nucleic Acids. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2017, 1861, 1371–1381. [Google Scholar] [CrossRef]
- Hoffmann, R.F.; Moshkin, Y.M.; Mouton, S.; Grzeschik, N.A.; Kalicharan, R.D.; Kuipers, J.; Wolters, A.H.G.; Nishida, K.; Romashchenko, A.V.; Postberg, J.; et al. Guanine Quadruplex Structures Localize to Heterochromatin. Nucleic Acids Res. 2015, 44, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.K.; Zybailov, B.L.; Maddukuri, L.; Gao, J.; Marecki, J.C.; Jaiswal, M.; Bell, M.R.; Griffin, W.C.; Reed, M.R.; Chib, S.; et al. Evidence That G-Quadruplex DNA Accumulates in the Cytoplasm and Participates in Stress Granule Assembly in Response to Oxidative Stress. J. Biol. Chem. 2016, 291, 18041–18057. [Google Scholar] [CrossRef] [Green Version]
- Lyonnais, S.; Gorelick, R.J.; Mergny, J.L.; le Cam, E.; Mirambeau, G. G-Quartets Direct Assembly of HIV-1 Nucleocapsid Protein along Single-Stranded DNA. Nucleic Acids Res. 2003, 31, 5754–5763. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Heaphy, S. Evidence for Interstrand Quadruplex Formation in the Dimerization of Human Immunodeficiency Virus 1 Genomic RNA. Proc. Natl. Acad. Sci. USA 1993, 90, 3393–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 Integrase Forms Stable Tetramers and Associates with LEDGF/P75 Protein in Human Cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; De Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/P75 Is Essential for Nuclear and Chromosomal Targeting of HIV-1 Integrase in Human Cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible Use of Nuclear Import Pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Brooks, T.A.; Hurley, L.H. The Role of Supercoiling in Transcriptional Control of MYC and Its Importance in Molecular Therapeutics. Nat. Rev. Cancer 2009, 9, 849–861. [Google Scholar] [CrossRef]
- Cogoi, S.; Shchekotikhin, A.E.; Xodo, L.E. HRAS Is Silenced by Two Neighboring G-Quadruplexes and Activated by MAZ, a Zinc-Finger Transcription Factor with DNA Unfolding Property. Nucleic Acids Res. 2014, 42, 8379–8388. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.-J.; Le, T.V.T.; Kim, K.; Hur, J.; Kim, K.K.; Park, H.-J. Novel Interaction of the Z-DNA Binding Domain of Human ADAR1 with the Oncogenic c-Myc Promoter G-Quadruplex. J. Mol. Biol. 2014, 426, 2594–2604. [Google Scholar] [CrossRef]
- Murat, P.; Balasubramanian, S. Existence and Consequences of G-Quadruplex Structures in DNA. Curr. Opin. Genet. Dev. 2014, 25, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Michelotti, G.A.; Michelotti, E.F.; Pullner, A.; Duncan, R.C.; Eick, D.; Levens, D. Multiple Single-Stranded Cis Elements Are Associated with Activated Chromatin of the Human c-Myc Gene in Vivo. Mol. Cell Biol. 1996, 16, 2656–2669. [Google Scholar] [CrossRef] [Green Version]
- Lam, E.Y.N.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. G-Quadruplex Structures Are Stable and Detectable in Human Genomic DNA. Nat. Commun. 2013, 4, 1796. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.K.; Dhar, S.; Henry, C.; Rich, A.; Ray, A. Epigenetic Regulation by Z-DNA Silencer Function Controls Cancer-Associated ADAM-12 Expression in Breast Cancer: Cross-Talk between MeCP2 and NF1 Transcription Factor Family. Cancer Res. 2013, 73, 736–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struhl, K.; Segal, E. Determinants of Nucleosome Positioning. Nat. Struct. Mol. Biol. 2013, 20, 267–273. [Google Scholar] [CrossRef]
- Sadeh, R.; Allis, C.D. Genome-Wide “Re”-Modeling of Nucleosome Positions. Cell 2011, 147, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.M.; Huppert, J.L. Stable G-Quadruplexes Are Found Outside Nucleosome-Bound Regions. Mol. Biosyst. 2009, 5, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, H. Enhancer-Promoter Interaction Facilitated by Transiently Forming G-Quadruplexes. Sci. Rep. 2015, 5, 9165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poeschla, E.M. Integrase, LEDGF/P75 and HIV Replication. Cell. Mol. Life Sci. 2008, 65, 1403–1424. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Wind-Rotolo, M.; Yang, H.-C.; Siliciano, J.D.; Siliciano, R.F. Experimental Approaches to the Study of HIV-1 Latency. Nat. Rev. Microbiol. 2007, 5, 95–106. [Google Scholar] [CrossRef]
- Bisgrove, D.; Lewinski, M.; Bushman, F.; Verdin, E. Molecular Mechanisms of HIV-1 Proviral Latency. Expert Rev. Anti-Infect. 2005, 3, 805–814. [Google Scholar] [CrossRef]
- Buffone, C.; Martinez-Lopez, A.; Fricke, T.; Opp, S.; Severgnini, M.; Cifola, I.; Petiti, L.; Frabetti, S.; Skorupka, K.; Zadrozny, K.K.; et al. Nup153 Unlocks the Nuclear Pore Complex for HIV-1 Nuclear Translocation in Nondividing Cells. J. Virol. 2018, 92, e00648-18. [Google Scholar] [CrossRef] [Green Version]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear Architecture Dictates HIV-1 Integration Site Selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Casartelli, N.; Pellin, D.; Rizzi, E.; Souque, P.; Severgnini, M.; Di Serio, C.; Fricke, T.; Diaz-Griffero, F.; Zimmer, C.; et al. Chromatin Organization at the Nuclear Pore Favours HIV Replication. Nat. Commun. 2015, 6, 6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Holde, K.; Zlatanova, J. Unusual DNA Structures, Chromatin and Transcription. Bioessays 1994, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Masai, H.; Kakusho, N.; Fukatsu, R.; Ma, Y.; Kanoh, Y.; Nagasawa, K. Molecular Architecture of G-Quadruplex Structures Generated on Duplex Rif1-Binding Sequences. J. Biol. Chem. 2018, 293, 17033–17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M.; Munroe, D.J. Weak Palindromic Consensus Sequences Are a Common Feature Found at the Integration Target Sites of Many Retroviruses. J. Virol. 2005, 79, 5211–5214. [Google Scholar] [CrossRef] [Green Version]
- Holman, A.G.; Coffin, J.M. Symmetrical Base Preferences Surrounding HIV-1, Avian Sarcoma/Leukosis Virus, and Murine Leukemia Virus Integration Sites. Proc. Natl. Acad. Sci. USA 2005, 102, 6103–6107. [Google Scholar] [CrossRef] [Green Version]
- Carteau, S.; Hoffmann, C.; Bushman, F.D. Chromosome Structure and HIV-1 CDNA Integration: Centromeric Alphoid Repeats Are a Disfavored Target. J. Virol. 1998, 72, 4005–4014. [Google Scholar] [CrossRef] [Green Version]
- Stevens, S.W.; Griffith, J.D. Sequence Analysis of the Human DNA Flanking Sites of Human Immunodeficiency Virus Type 1 Integration. J. Virol. 1996, 70, 6459–6462. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.P.; Ciuffi, A.; Leipzig, J.; Berry, C.C.; Bushman, F.D. HIV Integration Site Selection: Analysis by Massively Parallel Pyrosequencing Reveals Association with Epigenetic Modifications. Genome Res. 2007, 17, 1186–1194. [Google Scholar] [CrossRef] [Green Version]
- Derse, D.; Crise, B.; Li, Y.; Princler, G.; Lum, N.; Stewart, C.; McGrath, C.F.; Hughes, S.H.; Munroe, D.J.; Wu, X. Human T-Cell Leukemia Virus Type 1 Integration Target Sites in the Human Genome: Comparison with Those of Other Retroviruses. J. Virol. 2007, 81, 6731–6741. [Google Scholar] [CrossRef] [Green Version]
- Berry, C.; Hannenhalli, S.; Leipzig, J.; Bushman, F.D. Selection of Target Sites for Mobile DNA Integration in the Human Genome. PLoS Comput. Biol. 2006, 2, e157. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.D.W.; Huvet, M.; Melamed, A.; Maertens, G.N.; Bangham, C.R.M. Retroviruses Integrate into a Shared, Non-Palindromic DNA Motif. Nat. Microbiol. 2016, 2, 16212. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajoge, H.O.; Kohio, H.P.; Paparisto, E.; Coleman, M.D.; Wong, K.; Tom, S.K.; Bain, K.L.; Berry, C.C.; Arts, E.J.; Barr, S.D. G-Quadruplex DNA and Other Non-Canonical B-Form DNA Motifs Influence Productive and Latent HIV-1 Integration and Reactivation Potential. Viruses 2022, 14, 2494. https://doi.org/10.3390/v14112494
Ajoge HO, Kohio HP, Paparisto E, Coleman MD, Wong K, Tom SK, Bain KL, Berry CC, Arts EJ, Barr SD. G-Quadruplex DNA and Other Non-Canonical B-Form DNA Motifs Influence Productive and Latent HIV-1 Integration and Reactivation Potential. Viruses. 2022; 14(11):2494. https://doi.org/10.3390/v14112494
Chicago/Turabian StyleAjoge, Hannah O., Hinissan P. Kohio, Ermela Paparisto, Macon D. Coleman, Kemen Wong, Sean K. Tom, Katie L. Bain, Charles C. Berry, Eric J. Arts, and Stephen D. Barr. 2022. "G-Quadruplex DNA and Other Non-Canonical B-Form DNA Motifs Influence Productive and Latent HIV-1 Integration and Reactivation Potential" Viruses 14, no. 11: 2494. https://doi.org/10.3390/v14112494
APA StyleAjoge, H. O., Kohio, H. P., Paparisto, E., Coleman, M. D., Wong, K., Tom, S. K., Bain, K. L., Berry, C. C., Arts, E. J., & Barr, S. D. (2022). G-Quadruplex DNA and Other Non-Canonical B-Form DNA Motifs Influence Productive and Latent HIV-1 Integration and Reactivation Potential. Viruses, 14(11), 2494. https://doi.org/10.3390/v14112494