
Role of RNA Polymerase II Promoter-Proximal Pausing in Viral Transcription
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
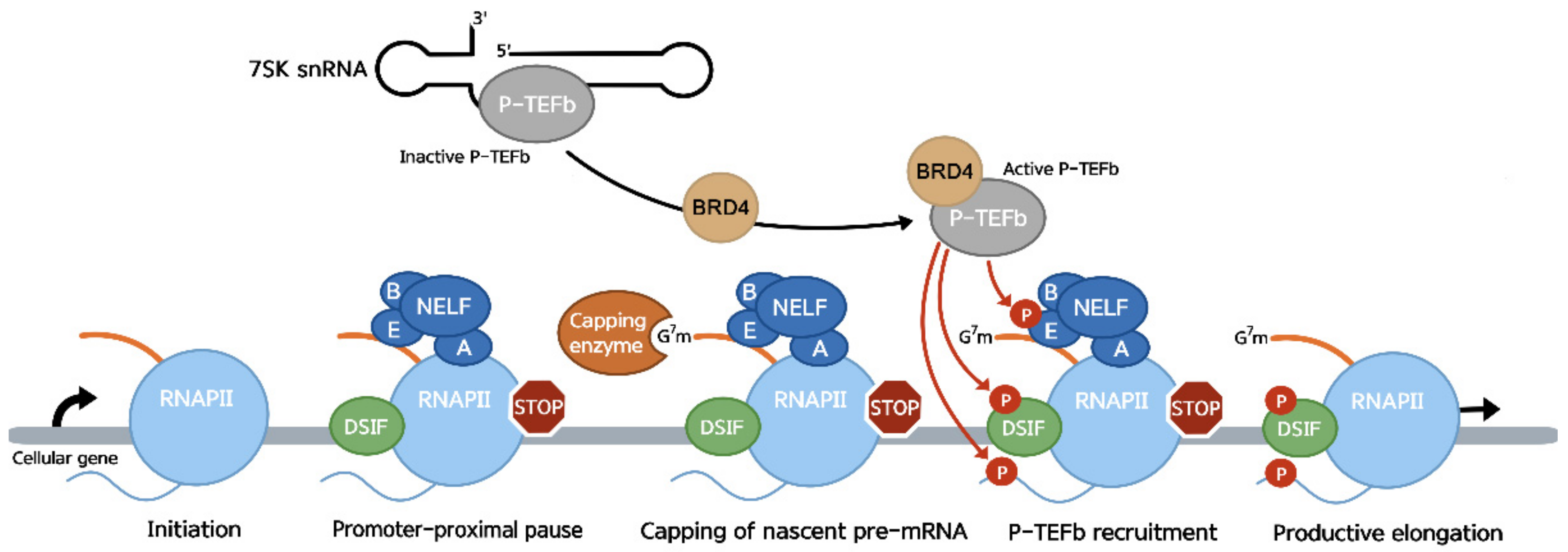
1. Promoter-Proximal Pausing during RNA Polymerase II Transcription
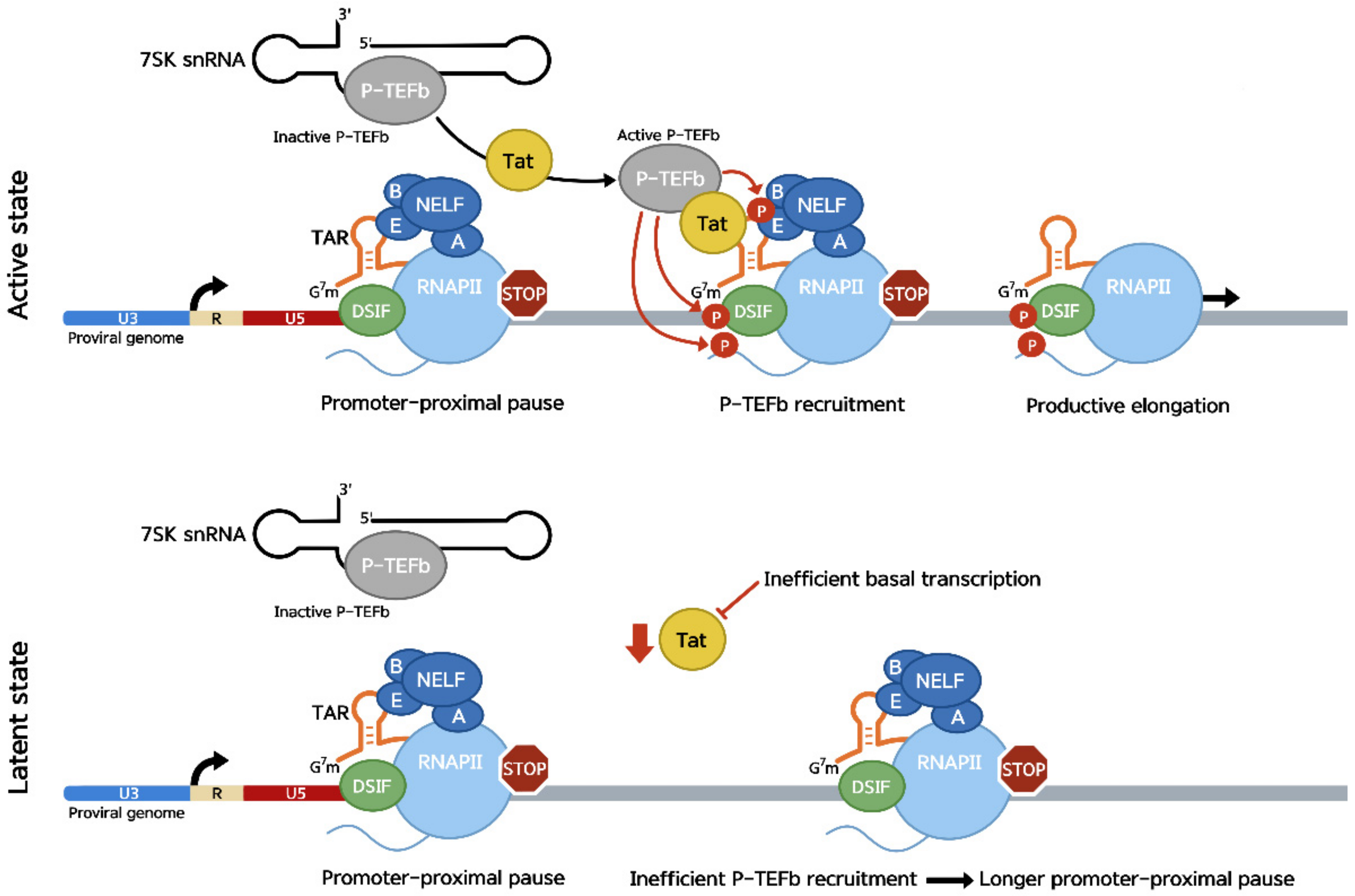
2. The Human Immunodeficiency Virus (HIV)
3. The Influenza A Virus (IAV)
4. The Hepatitis delta Virus (HDV)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.M.P.; van Schaik, F.M.A.; Varier, R.A.; Baltissen, M.P.A.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T.M. Selective Anchoring of TFIID to Nucleosomes by Trimethylation of Histone H3 Lysine 4. Cell 2007, 131, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, R.H.; Ladurner, A.G.; King, D.S.; Tjian, R. Structure and Function of a Human TAFII250 Double Bromodomain Module. Science 2000, 288, 1422–1425. [Google Scholar] [CrossRef]
- Thomas, M.C.; Chiang, C.-M. The General Transcription Machinery and General Cofactors. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 105–178. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Geiger, J.H.; Hahn, S.; Sigler, P.B. Crystal Structure of a Yeast TBP/TATA-Box Complex. Nature 1993, 365, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.H.; Hahn, S.; Lee, S.; Sigler, P.B. Crystal Structure of the Yeast TFIIA/TBP/DNA Complex. Science 1996, 272, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Hunziker, Y.; Sargent, D.F.; Richmond, T.J. Crystal Structure of a Yeast TFIIA/TBP/DNA Complex. Nature 1996, 381, 127–151. [Google Scholar] [CrossRef]
- Imbalzano, A.N.; Zaret, K.S.; Kingston, R.E. Transcription Factor (TF) IIB and TFIIA Can Independently Increase the Affinity of the TATA-Binding Protein for DNA. J. Biol. Chem. 1994, 269, 8280–8286. [Google Scholar] [CrossRef]
- Hieb, A.R.; Halsey, W.A.; Betterton, M.D.; Perkins, T.T.; Kugel, J.F.; Goodrich, J.A. TFIIA Changes the Conformation of the DNA in TBP/TATA Complexes and Increases Their Kinetic Stability. J. Mol. Biol. 2007, 372, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural Basis of Transcription Initiation by RNA Polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Louder, R.K.; Greber, B.J.; Grünberg, S.; Luo, J.; Fang, J.; Liu, Y.; Ranish, J.; Hahn, S.; Nogales, E. Structure of Human TFIID and Mechanism of TBP Loading onto Promoter DNA. Science 2018, 362, eaau8872. [Google Scholar] [CrossRef]
- Deng, W.; Roberts, S.G.E. TFIIB and the Regulation of Transcription by RNA Polymerase II. Chromosoma 2007, 116, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Taatjes, D.J. The Mediator Complex: A Central Integrator of Transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef]
- Fishburn, J.; Tomko, E.; Galburt, E.; Hahn, S. Double-Stranded DNA Translocase Activity of Transcription Factor TFIIH and the Mechanism of RNA Polymerase II Open Complex Formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3961–3966. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Shuman, S.; Lima, C.D. Structural Insights to How Mammalian Capping Enzyme Reads the CTD Code. Mol. Cell 2011, 43, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.J.; Trnka, M.J.; Bushnell, D.A.; Davis, R.E.; Mattei, P.-J.; Burlingame, A.L.; Kornberg, R.D. Structure of a Complete Mediator-RNA Polymerase II Pre-Initiation Complex. Cell 2016, 166, 1411–1422.e16. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, D.L.; Squazzo, S.L.; Muster, N.; Burckin, T.A.; Wachter, K.C.; Emigh, C.A.; McCleery, J.A.; Yates, J.R.; Hartzog, G.A. Dual Roles for Spt5 in Pre-MRNA Processing and Transcription Elongation Revealed by Identification of Spt5-Associated Proteins. Mol. Cell Biol. 2003, 23, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.S.; Chu, C.; Wada, T.; Handa, H.; Shatkin, A.J.; Reinberg, D. Functional Interactions of RNA-Capping Enzyme with Factors That Positively and Negatively Regulate Promoter Escape by RNA Polymerase II. Proc. Natl. Acad. Sci. USA 2004, 101, 7572–7577. [Google Scholar] [CrossRef]
- Wen, Y.; Shatkin, A.J. Transcription Elongation Factor HSPT5 Stimulates MRNA Capping. Genes Dev. 1999, 13, 1774–1779. [Google Scholar] [CrossRef]
- Narita, T.; Yung, T.M.C.; Yamamoto, J.; Tsuboi, Y.; Tanabe, H.; Tanaka, K.; Yamaguchi, Y.; Handa, H. NELF Interacts with CBC and Participates in 3′ End Processing of Replication-Dependent Histone MRNAs. Mol. Cell 2007, 26, 349–365. [Google Scholar] [CrossRef]
- Gilchrist, D.A.; dos Santos, G.; Fargo, D.C.; Xie, B.; Gao, Y.; Li, L.; Adelman, K. Pausing of RNA Polymerase II Disrupts DNA-Specified Nucleosome Organization to Enable Precise Gene Regulation. Cell 2010, 143, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, D.A.; Nechaev, S.; Lee, C.; Ghosh, S.K.B.; Collins, J.B.; Li, L.; Gilmour, D.S.; Adelman, K. NELF-Mediated Stalling of Pol II Can Enhance Gene Expression by Blocking Promoter-Proximal Nucleosome Assembly. Genes Dev. 2008, 22, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a Multisubunit Complex Containing RD, Cooperates with DSIF to Repress RNA Polymerase II Elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Inukai, N.; Narita, T.; Wada, T.; Handa, H. Evidence That Negative Elongation Factor Represses Transcription Elongation through Binding to a DRB Sensitivity-Inducing Factor/RNA Polymerase II Complex and RNA. Mol. Cell Biol. 2002, 22, 2918–2927. [Google Scholar] [CrossRef]
- Hartzog, G.A.; Fu, J. The Spt4-Spt5 Complex: A Multi-Faceted Regulator of Transcription Elongation. Biochim. Biophys. Acta 2013, 1829, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Gilmour, D.S. Identification of Regions in the Spt5 Subunit of DRB Sensitivity-Inducing Factor (DSIF) that Are Involved in Promoter-Proximal Pausing. J. Biol. Chem. 2017, 292, 5555–5570. [Google Scholar] [CrossRef]
- Narita, T.; Yamaguchi, Y.; Yano, K.; Sugimoto, S.; Chanarat, S.; Wada, T.; Kim, D.; Hasegawa, J.; Omori, M.; Inukai, N.; et al. Human Transcription Elongation Factor NELF: Identification of Novel Subunits and Reconstitution of the Functionally Active Complex. Mol. Cell Biol. 2003, 23, 1863–1873. [Google Scholar] [CrossRef]
- Pagano, J.M.; Kwak, H.; Waters, C.T.; Sprouse, R.O.; White, B.S.; Ozer, A.; Szeto, K.; Shalloway, D.; Craighead, H.G.; Lis, J.T. Defining NELF-E RNA Binding in HIV-1 and Promoter-Proximal Pause Regions. PLoS Genet. 2014, 10, e1004090. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the Elongation Phase of Transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK Small Nuclear RNA Inhibits the CDK9/Cyclin T1 Kinase to Control Transcription. Nature 2001, 414, 317–322. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK Small Nuclear RNA Binds to and Inhibits the Activity of CDK9/Cyclin T Complexes. Nature 2001, 414, 322–325. [Google Scholar] [CrossRef]
- Yik, J.H.N.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/Cyclin T) Kinase and RNA Polymerase II Transcription by the Coordinated Actions of HEXIM1 and 7SK SnRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Lu, X.; Zhu, X.; Li, Y.; Liu, M.; Yu, B.; Wang, Y.; Rao, M.; Yang, H.; Zhou, K.; Wang, Y.; et al. Multiple P-TEFbs Cooperatively Regulate the Release of Promoter-Proximally Paused RNA Polymerase II. Nucleic Acids Res. 2016, 44, 6853–6867. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of Human Immunodeficiency Virus Transcription: P-TEFb Phosphorylates RD and Dissociates Negative Effectors from the Transactivation Response Element. Mol. Cell Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shibata, H.; Handa, H. Transcription Elongation Factors DSIF and NELF: Promoter-Proximal Pausing and Beyond. Biochim. Biophys. Acta 2013, 1829, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Choe, H.; Farzan, M.; Sun, Y.; Sullivan, N.; Rollins, B.; Ponath, P.D.; Wu, L.; Mackay, C.R.; LaRosa, G.; Newman, W.; et al. The Beta-Chemokine Receptors CCR3 and CCR5 Facilitate Infection by Primary HIV-1 Isolates. Cell 1996, 85, 1135–1148. [Google Scholar] [CrossRef]
- Dalgleish, A.G.; Beverley, P.C.L.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; Weiss, R.A. The CD4 (T4) Antigen Is an Essential Component of the Receptor for the AIDS Retrovirus. Nature 1984, 312, 763–767. [Google Scholar] [CrossRef]
- Kozak, S.L.; Platt, E.J.; Madani, N.; Ferro, F.E.; Peden, K.; Kabat, D. CD4, CXCR-4, and CCR-5 Dependencies for Infections by Primary Patient and Laboratory-Adapted Isolates of Human Immunodeficiency Virus Type 1. J. Virol. 1997, 71, 873–882. [Google Scholar] [CrossRef]
- Valentin, A.; Albert, J.; Fenyö, E.M.; Asjö, B. Dual Tropism for Macrophages and Lymphocytes Is a Common Feature of Primary Human Immunodeficiency Virus Type 1 and 2 Isolates. J. Virol. 1994, 68, 6684–6689. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early Establishment of a Pool of Latently Infected, Resting CD4(+) T Cells during Primary HIV-1 Infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an Inducible HIV-1 Latent Reservoir during Highly Active Antiretroviral Therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef]
- Chun, T.W.; Fauci, A.S. Latent Reservoirs of HIV: Obstacles to the Eradication of Virus. Proc. Natl. Acad. Sci. USA 1999, 96, 10958–10961. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for Viral Persistence in the Presence of Antiviral Immune Responses and Antiretroviral Therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.N.; Singh, P.K. Cellular and Molecular Mechanisms of HIV-1 Integration Targeting. Cell Mol. Life Sci. 2018, 75, 2491–2507. [Google Scholar] [CrossRef]
- Schröder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Benleulmi, M.S.; Matysiak, J.; Henriquez, D.R.; Vaillant, C.; Lesbats, P.; Calmels, C.; Naughtin, M.; Leon, O.; Skalka, A.M.; Ruff, M.; et al. Intasome Architecture and Chromatin Density Modulate Retroviral Integration into Nucleosome. Retrovirology 2015, 12, 13. [Google Scholar] [CrossRef]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular Mechanisms of HIV Latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef]
- Jones, K.A.; Kadonaga, J.T.; Luciw, P.A.; Tjian, R. Activation of the AIDS Retrovirus Promoter by the Cellular Transcription Factor, Sp1. Science 1986, 232, 755–759. [Google Scholar] [CrossRef]
- Rittner, K.; Churcher, M.J.; Gait, M.J.; Karn, J. The Human Immunodeficiency Virus Long Terminal Repeat Includes a Specialised Initiator Element Which Is Required for Tat-Responsive Transcription. J. Mol. Biol. 1995, 248, 562–580. [Google Scholar] [CrossRef]
- Ross, E.K.; Buckler-White, A.J.; Rabson, A.B.; Englund, G.; Martin, M.A. Contribution of NF-Kappa B and Sp1 Binding Motifs to the Replicative Capacity of Human Immunodeficiency Virus Type 1: Distinct Patterns of Viral Growth Are Determined by T-Cell Types. J. Virol. 1991, 65, 4350–4358. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, M.; Schiralli Lester, G.M.; Lee, C.; Missra, A.; Wasserman, G.A.; Steffen, M.; Gilmour, D.S.; Henderson, A.J. Negative Elongation Factor (NELF) Coordinates RNA Polymerase II Pausing, Premature Termination, and Chromatin Remodeling to Regulate HIV Transcription. J. Biol. Chem. 2013, 288, 25995–26003. [Google Scholar] [CrossRef] [PubMed]
- Ping, Y.H.; Rana, T.M. DSIF and NELF Interact with RNA Polymerase II Elongation Complex and HIV-1 Tat Stimulates P-TEFb-Mediated Phosphorylation of RNA Polymerase II and DSIF during Transcription Elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef]
- Zhang, Z.; Klatt, A.; Gilmour, D.S.; Henderson, A.J. Negative Elongation Factor NELF Represses Human Immunodeficiency Virus Transcription by Pausing the RNA Polymerase II Complex. J. Biol. Chem. 2007, 282, 16981–16988. [Google Scholar] [CrossRef] [PubMed]
- Jakobovits, A.; Smith, D.H.; Jakobovits, E.B.; Capon, D.J. A Discrete Element 3′ of Human Immunodeficiency Virus 1 (HIV-1) and HIV-2 MRNA Initiation Sites Mediates Transcriptional Activation by an HIV Trans Activator. Mol. Cell. Biol. 1988, 8, 2555–2561. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Saifuddin, M. Regulation of HIV-1 Transcription. Gene Expr. 1999, 8, 67–84. [Google Scholar]
- Selby, M.J.; Bain, E.S.; Luciw, P.A.; Peterlin, B.M. Structure, Sequence, and Position of the Stem-Loop in Tar Determine Transcriptional Elongation by Tat through the HIV-1 Long Terminal Repeat. Genes Dev. 1989, 3, 547–558. [Google Scholar] [CrossRef]
- Muesing, M.A.; Smith, D.H.; Capon, D.J. Regulation of MRNA Accumulation by a Human Immunodeficiency Virus Trans-Activator Protein. Cell 1987, 48, 691–701. [Google Scholar] [CrossRef]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A Novel CDK9-Associated C-Type Cyclin Interacts Directly with HIV-1 Tat and Mediates Its High-Affinity, Loop-Specific Binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Mbonye, U.R.; Wang, B.; Gokulrangan, G.; Chance, M.R.; Karn, J. Phosphorylation of HEXIM1 at Tyr271 and Tyr274 Promotes Release of P-TEFb from the 7SK SnRNP Complex and Enhances Proviral HIV Gene Expression. Proteomics 2015, 15, 2078–2086. [Google Scholar] [CrossRef]
- Mancebo, H.S.; Lee, G.; Flygare, J.; Tomassini, J.; Luu, P.; Zhu, Y.; Peng, J.; Blau, C.; Hazuda, D.; Price, D.; et al. P-TEFb Kinase Is Required for HIV Tat Transcriptional Activation in Vivo and in Vitro. Genes Dev. 1997, 11, 2633–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.K.; Bourgeois, C.F.; Pearson, R.; Tyagi, M.; West, M.J.; Wong, J.; Wu, S.-Y.; Chiang, C.-M.; Karn, J. Recruitment of TFIIH to the HIV LTR Is a Rate-Limiting Step in the Emergence of HIV from Latency. EMBO J. 2006, 25, 3596–3604. [Google Scholar] [CrossRef]
- Kuzmina, A.; Krasnopolsky, S.; Taube, R. Super Elongation Complex Promotes Early HIV Transcription and Its Function Is Modulated by P-TEFb. Transcription 2017, 8, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Sierra, S.; Kupfer, B.; Kaiser, R. Basics of the Virology of HIV-1 and Its Replication. J. Clin. Virol. 2005, 34, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, L.S.; Dar, R.D.; Simpson, M.L. Transient-Mediated Fate Determination in a Transcriptional Circuit of HIV. Nat. Genet. 2008, 40, 466–470. [Google Scholar] [CrossRef]
- Duh, E.J.; Maury, W.J.; Folks, T.M.; Fauci, A.S.; Rabson, A.B. Tumor Necrosis Factor Alpha Activates Human Immunodeficiency Virus Type 1 through Induction of Nuclear Factor Binding to the NF-Kappa B Sites in the Long Terminal Repeat. Proc. Natl. Acad. Sci. USA 1989, 86, 5974–5978. [Google Scholar] [CrossRef]
- Nabel, G.; Baltimore, D. An Inducible Transcription Factor Activates Expression of Human Immunodeficiency Virus in T Cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Tong-Starksen, S.E.; Luciw, P.A.; Peterlin, B.M. Human Immunodeficiency Virus Long Terminal Repeat Responds to T-Cell Activation Signals. Proc. Natl. Acad. Sci. USA 1987, 84, 6845–6849. [Google Scholar] [CrossRef]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional Interference Antagonizes Proviral Gene Expression to Promote HIV Latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef]
- Winslow, B.J.; Pomerantz, R.J.; Bagasra, O.; Trono, D. HIV-1 Latency Due to the Site of Proviral Integration. Virology 1993, 196, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Rouzine, I.M.; Weinberger, A.D.; Weinberger, L.S. An Evolutionary Role for HIV Latency in Enhancing Viral Transmission. Cell 2015, 160, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M.I.; Stanwick, T.L.; Dempsey, M.P.; Stevenson, M. Quiescent T Lymphocytes as an Inducible Virus Reservoir in HIV-1 Infection. Science 1991, 254, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Mbonye, U.; Hokello, J.; Karn, J. T-Cell Receptor Signaling Enhances Transcriptional Elongation from Latent HIV Proviruses by Activating P-TEFb through an ERK-Dependent Pathway. J. Mol. Biol. 2011, 410, 896–916. [Google Scholar] [CrossRef] [PubMed]
- Spina, C.A.; Guatelli, J.C.; Richman, D.D. Establishment of a Stable, Inducible Form of Human Immunodeficiency Virus Type 1 DNA in Quiescent CD4 Lymphocytes in Vitro. J. Virol. 1995, 69, 2977–2988. [Google Scholar] [CrossRef]
- Beaton, A.R.; Krug, R.M. Selected Host Cell Capped RNA Fragments Prime Influenza Viral RNA Transcription in Vivo. Nucleic Acids Res. 1981, 9, 4423–4436. [Google Scholar] [CrossRef]
- Caton, A.J.; Robertson, J.S. Structure of the Host-Derived Sequences Present at the 5′ Ends of Influenza Virus MRNA. Nucleic Acids Res. 1980, 8, 2591–2603. [Google Scholar] [CrossRef]
- Dhar, R.; Chanock, R.M.; Lai, C.J. Nonviral Oligonucleotides at the 5′ Terminus of Cytoplasmic Influenza Viral MRNA Deduced from Cloned Complete Genomic Sequences. Cell 1980, 21, 495–500. [Google Scholar] [CrossRef]
- Fodor, E. The RNA Polymerase of Influenza a Virus: Mechanisms of Viral Transcription and Replication. Acta Virol. 2013, 57, 113–122. [Google Scholar] [CrossRef]
- Hatada, E.; Hasegawa, M.; Mukaigawa, J.; Shimizu, K.; Fukuda, R. Control of Influenza Virus Gene Expression: Quantitative Analysis of Each Viral RNA Species in Infected Cells. J. Biochem. 1989, 105, 537–546. [Google Scholar] [CrossRef]
- Mark, G.E.; Taylor, J.M.; Broni, B.; Krug, R.M. Nuclear Accumulation of Influenza Viral RNA Transcripts and the Effects of Cycloheximide, Actinomycin D., and Alpha-Amanitin. J. Virol. 1979, 29, 744–752. [Google Scholar] [CrossRef] [Green Version]
- Scholtissek, C.; Rott, R. Synthesis in Vivo of Influenza Virus plus and Minus Strand RNA and Its Preferential Inhibition by Antibiotics. Virology 1970, 40, 989–996. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Gurney, T., Jr.; Krug, R.M. Influenza Virus Gene Expression: Control Mechanisms at Early and Late Times of Infection and Nuclear-Cytoplasmic Transport of Virus-Specific RNAs. J. Virol. 1987, 61, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Illmensee, R.; Litwin, S.; Herring, L.; Broni, B.; Krug, R.M. Use of Specific Radioactive Probes to Study Transcription and Replication of the Influenza Virus Genome. J. Virol. 1977, 21, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural Insights into RNA Synthesis by the Influenza Virus Transcription-Replication Machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef]
- McGeoch, D.; Fellner, P.; Newton, C. Influenza Virus Genome Consists of Eight Distinct RNA Species. Proc. Natl. Acad. Sci. USA 1976, 73, 3045–3049. [Google Scholar] [CrossRef]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Hsu, M.T.; Parvin, J.D.; Gupta, S.; Krystal, M.; Palese, P. Genomic RNAs of Influenza Viruses Are Held in a Circular Conformation in Virions and in Infected Cells by a Terminal Panhandle. Proc. Natl. Acad. Sci. USA 1987, 84, 8140–8144. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of Influenza A Polymerase Bound to the Viral RNA Promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef]
- Argos, P. A Sequence Motif in Many Polymerases. Nucleic Acids Res. 1988, 16, 9909–9916. [Google Scholar] [CrossRef]
- Biswas, S.K.; Nayak, D.P. Mutational Analysis of the Conserved Motifs of Influenza A Virus Polymerase Basic Protein 1. J. Virol. 1994, 68, 1819–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braam, J.; Ulmanen, I.; Krug, R.M. Molecular Model of a Eucaryotic Transcription Complex: Functions and Movements of Influenza P Proteins during Capped RNA-Primed Transcription. Cell 1983, 34, 609–618. [Google Scholar] [CrossRef]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of Four Conserved Motifs among the RNA-Dependent Polymerase Encoding Elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar] [CrossRef] [PubMed]
- Arranz, R.; Coloma, R.; Chichón, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortín, J.; Martín-Benito, J. The Structure of Native Influenza Virion Ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Blaas, D.; Patzelt, E.; Kuechler, E. Cap-Recognizing Protein of Influenza Virus. Virology 1982, 116, 339–348. [Google Scholar] [CrossRef]
- Dias, A.; Bouvier, D.; Crépin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W.H. The Cap-Snatching Endonuclease of Influenza Virus Polymerase Resides in the PA Subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef]
- Fechter, P.; Mingay, L.; Sharps, J.; Chambers, A.; Fodor, E.; Brownlee, G.G. Two Aromatic Residues in the PB2 Subunit of Influenza A RNA Polymerase Are Crucial for Cap Binding. J. Biol. Chem. 2003, 278, 20381–20388. [Google Scholar] [CrossRef]
- Fechter, P.; Brownlee, G.G. Recognition of MRNA Cap Structures by Viral and Cellular Proteins. J. Gen. Virol. 2005, 86, 1239–1249. [Google Scholar] [CrossRef]
- Guilligay, D.; Tarendeau, F.; Resa-Infante, P.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.H.H.; Ortin, J.; Hart, D.J.; et al. The Structural Basis for Cap Binding by Influenza Virus Polymerase Subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef]
- Hara, K.; Schmidt, F.I.; Crow, M.; Brownlee, G.G. Amino Acid Residues in the N-Terminal Region of the PA Subunit of Influenza A Virus RNA Polymerase Play a Critical Role in Protein Stability, Endonuclease Activity, Cap Binding, and Virion RNA Promoter Binding. J. Virol. 2006, 80, 7789–7798. [Google Scholar] [CrossRef]
- Penn, C.R.; Blaas, D.; Kuechler, E.; Mahy, B.W.J. Identification of the Cap-Binding Protein of Two Strains of Influenza A/FPA. J. Gen. Virol. 1982, 62, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Ulmanen, I.; Broni, B.A.; Krug, R.M. Role of Two of the Influenza Virus Core P Proteins in Recognizing Cap 1 Structures (M7GpppNm) on RNAs and in Initiating Viral RNA Transcription. Proc. Natl. Acad. Sci. USA 1981, 78, 7355–7359. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Bartlam, M.; Lou, Z.; Chen, S.; Zhou, J.; He, X.; Lv, Z.; Ge, R.; Li, X.; Deng, T.; et al. Crystal Structure of an Avian Influenza Polymerase PA(N) Reveals an Endonuclease Active Site. Nature 2009, 458, 909–913. [Google Scholar] [CrossRef]
- Biswas, S.K.; Boutz, P.L.; Nayak, D.P. Influenza Virus Nucleoprotein Interacts with Influenza Virus Polymerase Proteins. J. Virol. 1998, 72, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Elton, D.; Medcalf, L.; Digard, P. Functional Domains of the Influenza A Virus PB2 Protein: Identification of NP- and PB1-Binding Sites. Virology 2004, 321, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Ishihama, A.; Nagata, K. Reconstitution of Influenza Virus RNA-Nucleoprotein Complexes Structurally Resembling Native Viral Ribonucleoprotein Cores. J. Biol. Chem. 1990, 265, 11151–11155. [Google Scholar] [CrossRef]
- De Vlugt, C.; Sikora, D.; Pelchat, M. Insight into Influenza: A Virus Cap-Snatching. Viruses 2018, 10, 641. [Google Scholar] [CrossRef]
- Engelhardt, O.G.; Smith, M.; Fodor, E. Association of the Influenza A Virus RNA-Dependent RNA Polymerase with Cellular RNA Polymerase II. J. Virol. 2005, 79, 5812–5818. [Google Scholar] [CrossRef]
- Walker, A.P.; Fodor, E. Interplay between Influenza Virus and the Host RNA Polymerase II Transcriptional Machinery. Trends Microbiol. 2019, 27, 398–407. [Google Scholar] [CrossRef]
- Martínez-Alonso, M.; Hengrung, N.; Fodor, E. RNA-Free and Ribonucleoprotein-Associated Influenza Virus Polymerases Directly Bind the Serine-5-Phosphorylated Carboxyl-Terminal Domain of Host RNA Polymerase II. J. Virol. 2016, 90, 6014–6021. [Google Scholar] [CrossRef]
- Geerts-Dimitriadou, C.; Goldbach, R.; Kormelink, R. Preferential Use of RNA Leader Sequences during Influenza A Transcription Initiation in Vivo. Virology 2011, 409, 27–32. [Google Scholar] [CrossRef]
- Shi, L.; Summers, D.F.; Peng, Q.; Galarz, J.M. Influenza A Virus RNA Polymerase Subunit PB2 Is the Endonuclease Which Cleaves Host Cell MRNA and Functions Only as the Trimeric Enzyme. Virology 1995, 208, 38–47. [Google Scholar] [CrossRef]
- Sikora, D.; Rocheleau, L.; Brown, E.G.; Pelchat, M. Deep Sequencing Reveals the Eight Facets of the Influenza A/HongKong/1/1968 (H3N2) Virus Cap-Snatching Process. Sci. Rep. 2014, 4, 6181. [Google Scholar] [CrossRef]
- Sikora, D.; Rocheleau, L.; Brown, E.G.; Pelchat, M. Influenza A Virus Cap-Snatches Host RNAs Based on Their Abundance Early after Infection. Virology 2017, 509, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Bouloy, M.; Morgan, M.A.; Shatkin, A.J.; Krug, R.M. Cap and Internal Nucleotides of Reovirus MRNA Primers Are Incorporated into Influenza Viral Complementary RNA during Transcription in Vitro. J. Virol. 1979, 32, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Bouloy, M.; Plotch, S.J.; Krug, R.M. Globin MRNAs Are Primers for the Transcription of Influenza Viral RNA in Vitro. Proc. Natl. Acad. Sci. USA 1978, 75, 4886–4890. [Google Scholar] [CrossRef] [PubMed]
- Plotch, S.J.; Bouloy, M.; Krug, R.M. Transfer of 5′-Terminal Cap of Globin MRNA to Influenza Viral Complementary RNA during Transcription in Vitro. Proc. Natl. Acad. Sci. USA 1979, 76, 1618–1622. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Rao, P.; Krug, R.M. The Active Sites of the Influenza Cap-Dependent Endonuclease Are on Different Polymerase Subunits. EMBO J. 2001, 20, 2078–2086. [Google Scholar] [CrossRef]
- Plotch, S.J.; Bouloy, M.; Ulmanen, I.; Krug, R.M. A Unique Cap(M7GpppXm)-Dependent Influenza Virion Endonuclease Cleaves Capped RNAs to Generate the Primers That Initiate Viral RNA Transcription. Cell 1981, 23, 847–858. [Google Scholar] [CrossRef]
- Robertson, H.D.; Dickson, E.; Plotch, S.J.; Krug, R.M. Identification of the RNA Region Transferred from a Representative Primer, β-globin MRNA, to Influenza MRNA during in Vitro Transcription. Nucleic Acids Res. 1980, 8, 925–942. [Google Scholar] [CrossRef]
- De Vlugt, C.; Sikora, D.; Rocheleau, L.; Pelchat, M. Priming and Realignment by the Influenza a Virus RdRp Is Dependent on the Length of the Host Primers and the Extent of Base Pairing to Viral RNA. Virology 2019, 536, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Geerts-Dimitriadou, C.; Zwart, M.P.; Goldbach, R.; Kormelink, R. Base-Pairing Promotes Leader Selection to Prime in Vitro Influenza Genome Transcription. Virology 2011, 409, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Hagen, M.; Chung, T.D.; Butcher, J.A.; Krystal, M. Recombinant Influenza Virus Polymerase: Requirement of Both 5′ and 3′ Viral Ends for Endonuclease Activity. J. Virol. 1994, 68, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Te Velthuis, A.J.W.; Oymans, J. Initiation, Elongation, and Realignment during Influenza Virus MRNA Synthesis. J. Virol. 2018, 92, e01775-17. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.Y.; Vreede, F.T.; Smith, M.; Engelhardt, O.G.; Fodor, E. Influenza Virus Inhibits RNA Polymerase II Elongation. Virology 2006, 351, 210–217. [Google Scholar] [CrossRef]
- Bauer, D.L.; Tellier, M.; Martínez-Alonso, M.; Nojima, T.; Proudfoot, N.J.; Murphy, S.; Fodor, E. Influenza Virus Mounts a Two-Pronged Attack on Host RNA Polymerase II Transcription. Cell Rep. 2018, 23, 2119–2129.e3. [Google Scholar] [CrossRef]
- Levene, R.E.; Gaglia, M.M. Host Shutoff in Influenza A Virus: Many Means to an End. Viruses 2018, 10, 475. [Google Scholar] [CrossRef]
- Koppstein, D.; Ashour, J.; Bartel, D.P. Sequencing the Cap-Snatching Repertoire of H1N1 Influenza Provides Insight into the Mechanism of Viral Transcription Initiation. Nucleic Acids Res. 2015, 43, 5052–5064. [Google Scholar] [CrossRef]
- Sureau, C.; Negro, F. The Hepatitis Delta Virus: Replication and Pathogenesis. J. Hepatol. 2016, 64, S102–S116. [Google Scholar] [CrossRef]
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, Sequence and Expression of the Hepatitis Delta (Delta) Viral Genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef]
- Taylor, J.; Pelchat, M. Origin of Hepatitis Delta Virus. Future Microbiol. 2010, 5, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.J.; Choo, Q.L.; Wang, K.S.; Govindarajan, S.; Redeker, A.G.; Gerin, J.L.; Houghton, M. A Single Antigenomic Open Reading Frame of the Hepatitis Delta Virus Encodes the Epitope(s) of Both Hepatitis Delta Antigen Polypeptides P24 Delta and P27 Delta. J. Virol. 1988, 62, 594–599. [Google Scholar] [CrossRef]
- Chang, F.L.; Chen, P.J.; Tu, S.J.; Wang, C.J.; Chen, D.S. The Large Form of Hepatitis Delta Antigen Is Crucial for Assembly of Hepatitis Delta Virus. Proc. Natl. Acad. Sci. USA 1991, 88, 8490–8494. [Google Scholar] [CrossRef]
- Modahl, L.E.; Lai, M.M. The Large Delta Antigen of Hepatitis Delta Virus Potently Inhibits Genomic but Not Antigenomic RNA Synthesis: A Mechanism Enabling Initiation of Viral Replication. J. Virol. 2000, 74, 7375–7380. [Google Scholar] [CrossRef] [PubMed]
- Moraleda, G.; Taylor, J. Host RNA Polymerase Requirements for Transcription of the Human Hepatitis Delta Virus Genome. J. Virol. 2001, 75, 10161–10169. [Google Scholar] [CrossRef] [PubMed]
- Gudima, S.; Wu, S.Y.; Chiang, C.M.; Moraleda, G.; Taylor, J. Origin of Hepatitis Delta Virus MRNA. J. Virol. 2000, 74, 7204–7210. [Google Scholar] [CrossRef]
- Nie, X.; Chang, J.; Taylor, J.M. Alternative Processing of Hepatitis Delta Virus Antigenomic RNA Transcripts. J. Virol. 2004, 78, 4517–4524. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.B.; Taylor, J. The RNAs of Hepatitis Delta Virus Are Copied by RNA Polymerase II in Nuclear Homogenates. J. Virol. 1993, 67, 6965–6972. [Google Scholar] [CrossRef]
- Abrahem, A.; Pelchat, M. Formation of an RNA Polymerase II Preinitiation Complex on an RNA Promoter Derived from the Hepatitis Delta Virus RNA Genome. Nucleic Acids Res. 2008, 36, 5201–5211. [Google Scholar] [CrossRef]
- Greco-Stewart, V.S.; Miron, P.; Abrahem, A.; Pelchat, M. The Human RNA Polymerase II Interacts with the Terminal Stem-Loop Regions of the Hepatitis Delta Virus RNA Genome. Virology 2007, 357, 68–78. [Google Scholar] [CrossRef]
- Li, Y.-J.; Macnaughton, T.; Gao, L.; Lai, M.M.C. RNA-Templated Replication of Hepatitis Delta Virus: Genomic and Antigenomic RNAs Associate with Different Nuclear Bodies. J. Virol. 2006, 80, 6478–6486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macnaughton, T.B.; Shi, S.T.; Modahl, L.E.; Lai, M.M.C. Rolling Circle Replication of Hepatitis Delta Virus RNA Is Carried Out by Two Different Cellular RNA Polymerases. J. Virol. 2002, 76, 3920–3927. [Google Scholar] [CrossRef] [PubMed]
- Modahl, L.E.; Macnaughton, T.B.; Zhu, N.; Johnson, D.L.; Lai, M.M.C. RNA-Dependent Replication and Transcription of Hepatitis Delta Virus RNA Involve Distinct Cellular RNA Polymerases. Mol. Cell Biol. 2000, 20, 6030–6039. [Google Scholar] [CrossRef] [PubMed]
- Greco-Stewart, V.S.; Schissel, E.; Pelchat, M. The Hepatitis Delta Virus RNA Genome Interacts with the Human RNA Polymerases I and III. Virology 2009, 386, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Nie, X.; Chang, H.E.; Han, Z.; Taylor, J. Transcription of Hepatitis Delta Virus RNA by RNA Polymerase II. J. Virol. 2008, 82, 1118–1127. [Google Scholar] [CrossRef]
- Filipovska, J.; Konarska, M.M. Specific HDV RNA-Templated Transcription by Pol II in Vitro. RNA 2000, 6, 41–54. [Google Scholar] [CrossRef]
- Chang, J.; Nie, X.; Gudima, S.; Taylor, J. Action of Inhibitors on Accumulation of Processed Hepatitis Delta Virus RNAs. J. Virol. 2006, 80, 3205–3214. [Google Scholar] [CrossRef]
- Beeharry, Y.; Rocheleau, L.; Pelchat, M. Conserved Features of an RNA Promoter for RNA Polymerase II Determined from Sequence Heterogeneity of a Hepatitis Delta Virus Population. Virology 2014, 450–451, 165–173. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Filipovska, J.; Yano, K.; Furuya, A.; Inukai, N.; Narita, T.; Wada, T.; Sugimoto, S.; Konarska, M.M.; Handa, H. Stimulation of RNA Polymerase II Elongation by Hepatitis Delta Antigen. Science 2001, 293, 124–127. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Mura, T.; Chanarat, S.; Okamoto, S.; Handa, H. Hepatitis Delta Antigen Binds to the Clamp of RNA Polymerase II and Affects Transcriptional Fidelity. Genes Cells 2007, 12, 863–875. [Google Scholar] [CrossRef]
- Nedialkov, Y.A.; Gong, X.Q.; Hovde, S.L.; Yamaguchi, Y.; Handa, H.; Geiger, J.H.; Yan, H.; Burton, Z.F. NTP-Driven Translocation by Human RNA Polymerase II. J. Biol. Chem. 2003, 278, 18303–18312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaborowska, J.; Baumli, S.; Laitem, C.; O’Reilly, D.; Thomas, P.H.; O’Hare, P.; Murphy, S. Herpes Simplex Virus 1 (HSV-1) ICP22 Protein Directly Interacts with Cyclin-Dependent Kinase (CDK)9 to Inhibit RNA Polymerase II Transcription Elongation. PLoS ONE 2014, 9, e107654. [Google Scholar] [CrossRef]
- Fraser, K.A.; Rice, S.A. Herpes Simplex Virus Immediate-Early Protein ICP22 Triggers Loss of Serine 2-Phosphorylated RNA Polymerase II. J. Virol. 2007, 81, 5091–5101. [Google Scholar] [CrossRef] [PubMed]
- Birkenheuer, C.H.; Baines, J.D. RNA Polymerase II Promoter-Proximal Pausing and Release to Elongation Are Key Steps Regulating Herpes Simplex Virus 1 Transcription. J. Virol. 2020, 94, e02035-19. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whelan, M.; Pelchat, M. Role of RNA Polymerase II Promoter-Proximal Pausing in Viral Transcription. Viruses 2022, 14, 2029. https://doi.org/10.3390/v14092029
Whelan M, Pelchat M. Role of RNA Polymerase II Promoter-Proximal Pausing in Viral Transcription. Viruses. 2022; 14(9):2029. https://doi.org/10.3390/v14092029
Chicago/Turabian StyleWhelan, Marilyn, and Martin Pelchat. 2022. "Role of RNA Polymerase II Promoter-Proximal Pausing in Viral Transcription" Viruses 14, no. 9: 2029. https://doi.org/10.3390/v14092029