
Evolutionary Conservation of PP2A Antagonism and G2/M Cell Cycle Arrest in Maedi-Visna Virus Vif

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Cloning
2.2. RNA Isolation, cDNA Synthesis, and Inhibitor Treatments
2.3. Propidium Iodide Staining and Cell Cycle Arrest
2.4. Fluorescence Microscopy
2.5. Flow Cytometry
2.6. Statistical Analyses and MVV Vif Modeling
3. Results
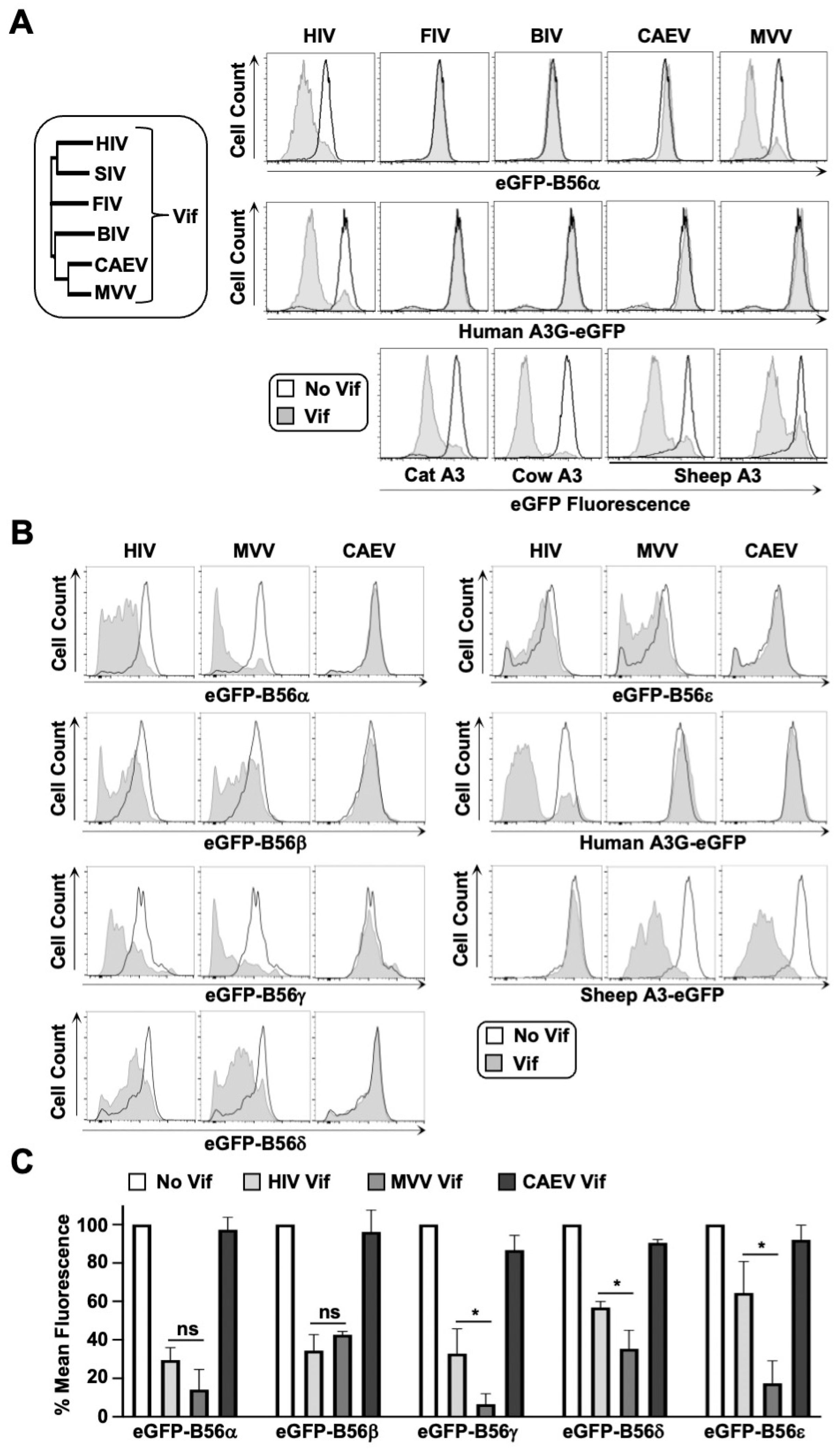
3.1. B56α-ε Degradation Activity Is a Conserved Function of MVV Vif
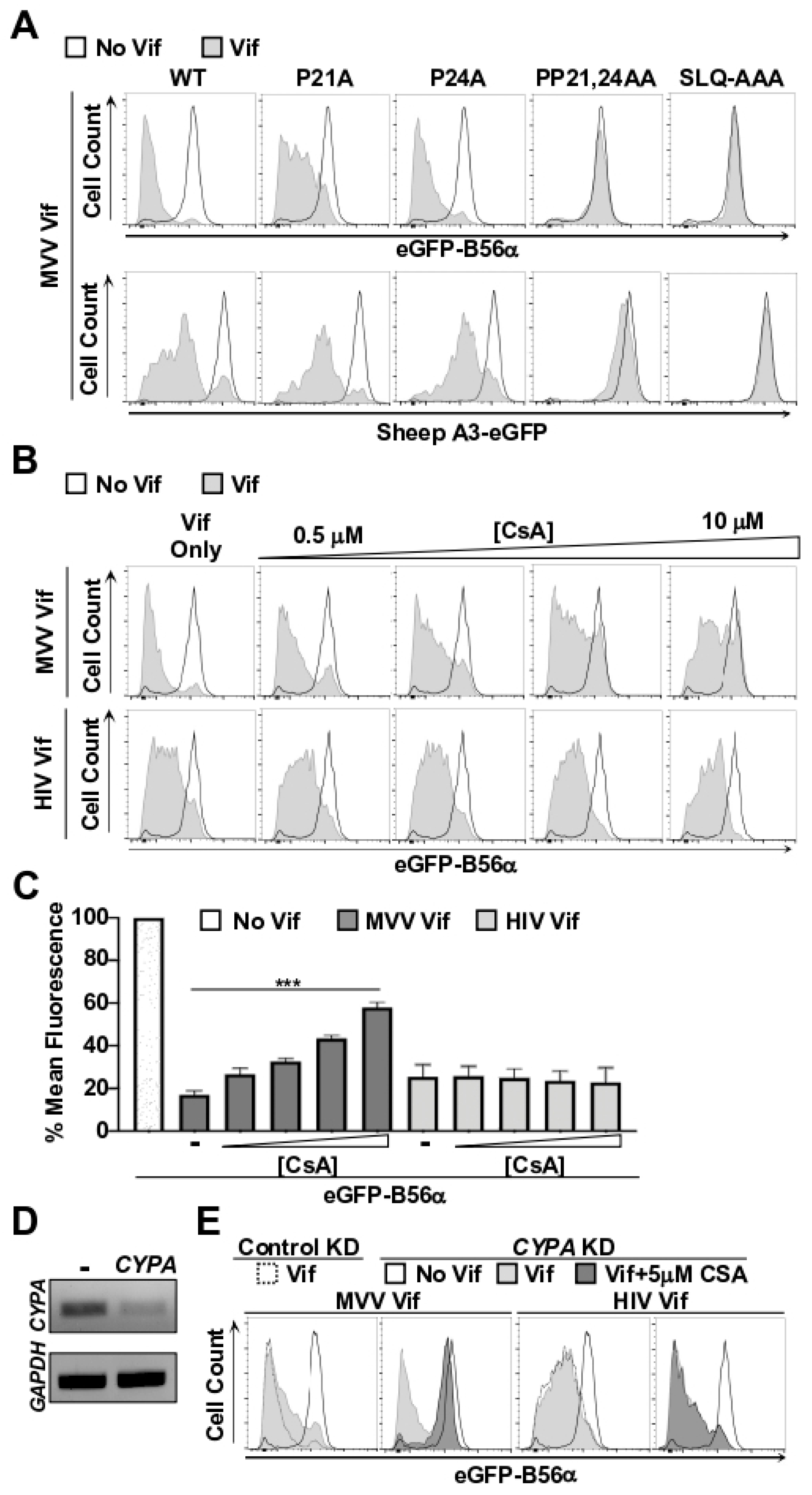
3.2. CYPA Is Required for MVV Vif-Induced Degradation of B56 Proteins
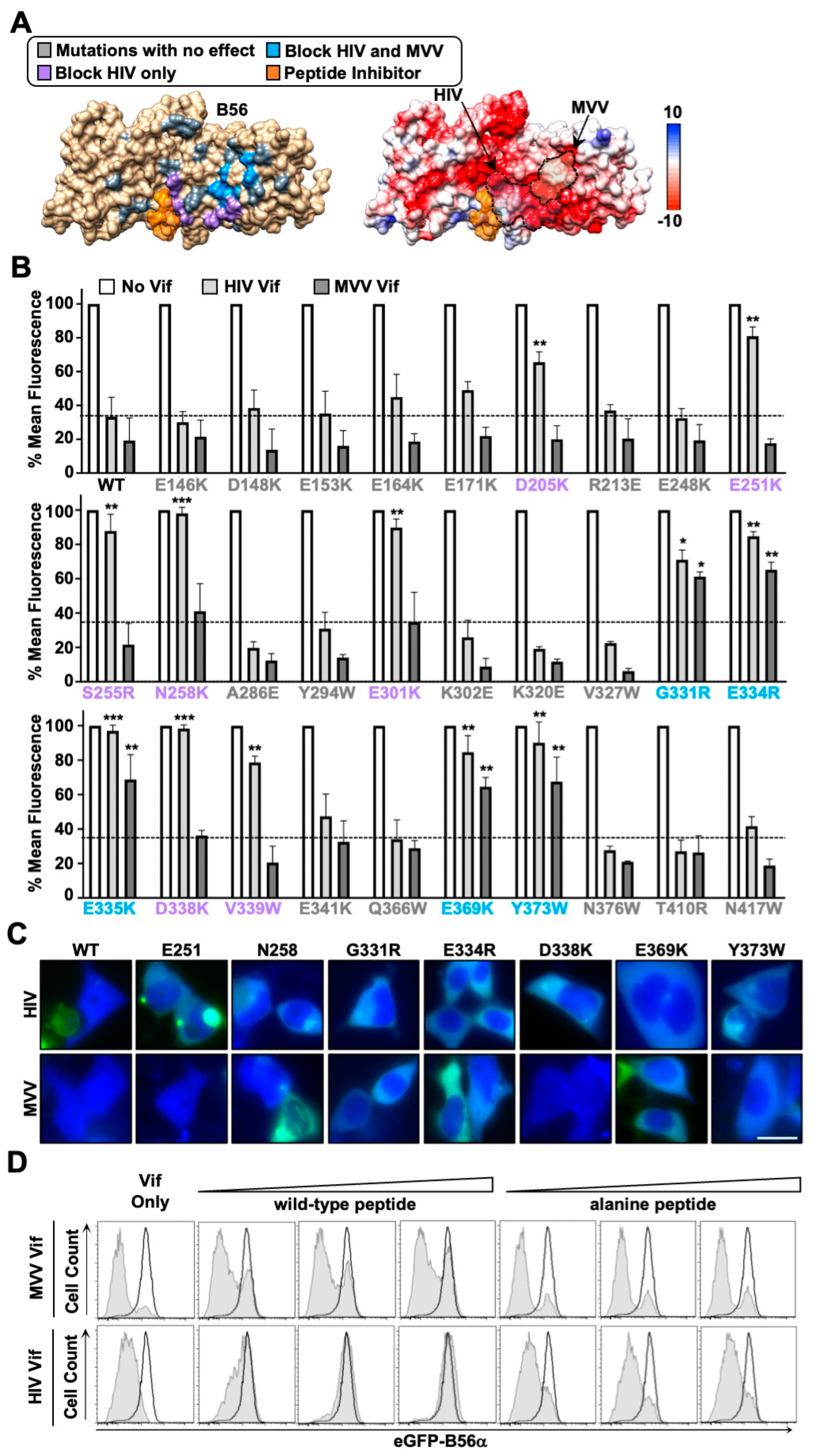
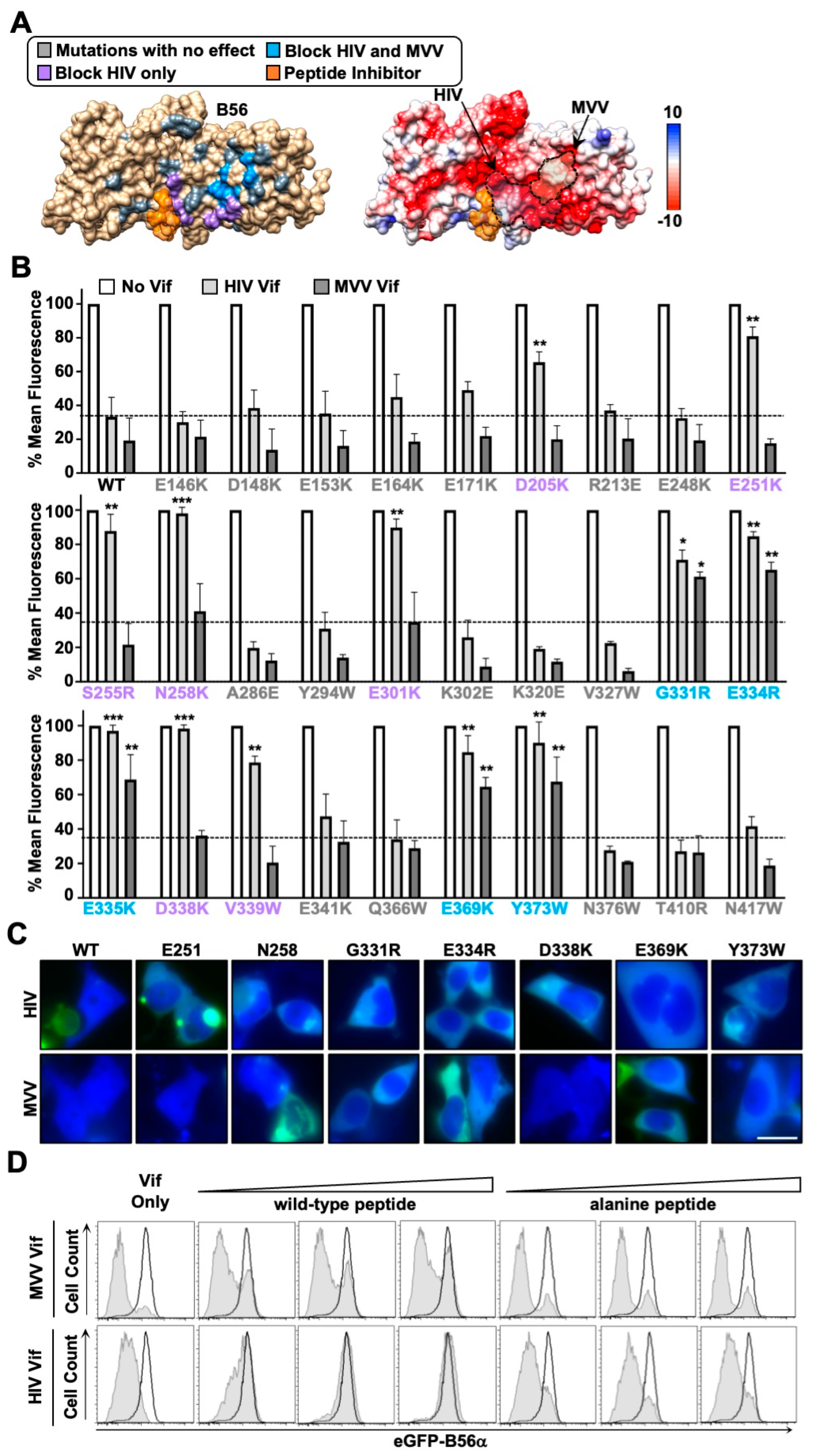
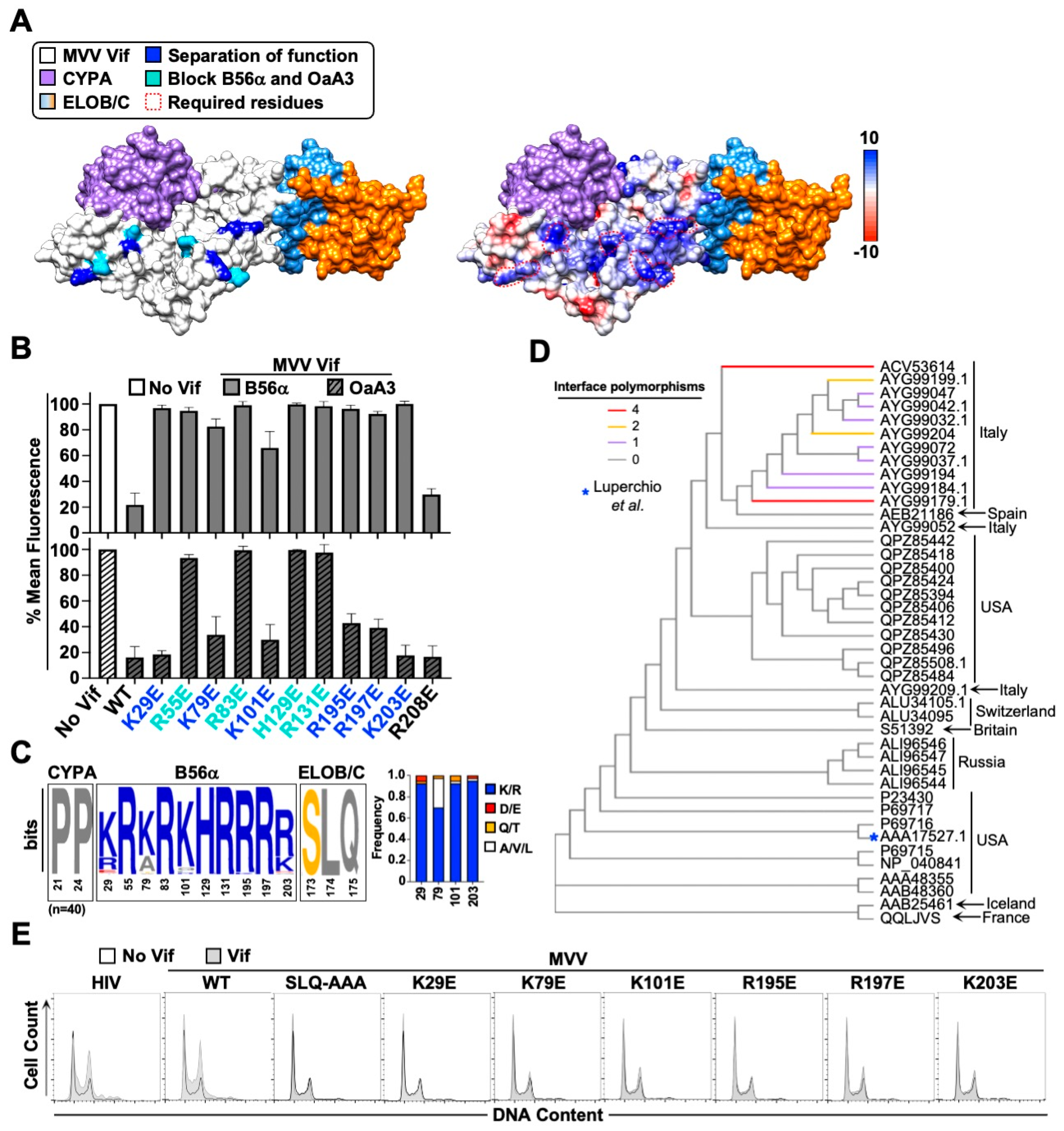
3.3. HIV and MVV Vif Bind Partially Distinct B56 Surfaces through Clustered Electrostatic Interactions
3.4. G2/M Cell Cycle Arrest and Conservation of the MVV Vif-B56 Interface
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Harris, R.; Dudley, J. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins--ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, V.B.N.; Landau, N. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol. 2015, 16, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking CBF-β and CUL5 E3 ligase complex by HIV-1 Vif. Nature 2014, 505, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, E.J.; Matheson, N.J.; Wals, K.; van den Boomen, D.J.; Antrobus, R.; Williamson, J.C.; Lehner, P.J. Temporal proteomic analysis of HIV infection reveals remodelling of the host phosphoproteome by lentiviral Vif variants. Elife 2016, 5, e18296. [Google Scholar] [CrossRef]
- Naamati, A.; Williamson, J.C.; Greenwood, E.J.; Marelli, S.; Lehner, P.J.; Matheson, N.J. Functional proteomic atlas of HIV infection in primary human CD4+ T cells. Elife 2019, 8, e41431. [Google Scholar] [CrossRef] [PubMed]
- McCright, B.; Rivers, A.M.; Audlin, S.; Virshup, D.M. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J. Biol. Chem. 1996, 271, 22081–22089. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Bajaj, R.; Bollen, M.; Peti, W.; Page, R. Expanding the PP2A Interactome by Defining a B56-Specific SLiM. Structure 2016, 24, 2174–2181. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J. Protein phosphatases in the regulation of mitosis. J. Cell Biol. 2019, 218, 395–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, Z.; Yu, T.; Yang, H.; Virshup, D.M.; Kops, G.J.; Lee, S.H.; Zhou, W.; Li, X.; Xu, W.; et al. Crystal structure of a PP2A B56-BubR1 complex and its implications for PP2A substrate recruitment and localization. Protein Cell 2016, 7, 516–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salamango, D.J.; Ikeda, T.; Moghadasi, S.A.; Wang, J.; McCann, J.L.; Serebrenik, A.A.; Ebrahimi, D.; Jarvis, M.C.; Brown, W.L.; Harris, R.S. HIV-1 Vif Triggers Cell Cycle Arrest by Degrading Cellular PPP2R5 Phospho-regulators. Cell Rep. 2019, 29, 1057–1065.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marelli, S.; Williamson, J.C.; Protasio, A.V.; Naamati, A.; Greenwood, E.J.; Deane, J.E.; Lehner, P.J.; Matheson, N.J. Antagonism of PP2A is an independent and conserved function of HIV-1 Vif and causes cell cycle arrest. Elife 2020, 9, e53036. [Google Scholar] [CrossRef] [Green Version]
- Izumi, T.; Io, K.; Matsui, M.; Shirakawa, K.; Shinohara, M.; Nagai, Y.; Kawahara, M.; Kobayashi, M.; Kondoh, H.; Misawa, N.; et al. HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. USA 2010, 107, 20798–20803. [Google Scholar] [CrossRef] [Green Version]
- DeHart, J.L.; Bosque, A.; Harris, R.S.; Planelles, V. Human immunodeficiency virus type 1 Vif induces cell cycle delay via recruitment of the same E3 ubiquitin ligase complex that targets APOBEC3 proteins for degradation. J. Virol. 2008, 82, 9265–9272. [Google Scholar] [CrossRef] [Green Version]
- Nagata, K.; Shindo, K.; Matsui, Y.; Shirakawa, K.; Takaori-Kondo, A. Critical role of PP2A-B56 family protein degradation in HIV-1 Vif mediated G2 cell cycle arrest. Biochem. Biophys. Res. Commun. 2020, 527, 257–263. [Google Scholar] [CrossRef]
- Salamango, D.J.; Harris, R.S. Dual Functionality of HIV-1 Vif in APOBEC3 Counteraction and Cell Cycle Arrest. Front. Microbiol. 2020, 11, 622012. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Z.; Huan, C.; Wang, H.; Su, X.; Zhang, W. CAEV Vif Hijacks ElonginB/C, CPYA and Cullin5 to Assemble the E3 Ubiquitin Ligase Complex Stepwise to Degrade oaA3Z2-Z3. Front. Microbiol. 2019, 10, 565. [Google Scholar] [CrossRef]
- Salamango, D.; McCann, J.; Demir, O.; Becker, J.; Wang, J.; Lingappa, J.; Temiz, N.; Brown, W.; Amaro, R.; Harris, R. Functional and structural insights into a Vif/PPP2R5 complex elucidated using patient HIV-1 isolates and computational modeling. J. Virol. 2020, 94, e00631-20. [Google Scholar] [CrossRef]
- Kane, J.R.; Stanley, D.J.; Hultquist, J.F.; Johnson, J.R.; Mietrach, N.; Binning, J.M.; Jónsson, S.R.; Barelier, S.; Newton, B.W.; Johnson, T.L.; et al. Lineage-Specific Viral Hijacking of Non-canonical E3 Ubiquitin Ligase Cofactors in the Evolution of Vif Anti-APOBEC3 Activity. Cell Rep. 2015, 11, 1236–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, K.M.; Hu, Y.; Rubene, D.; Cook, M.; Ziegler, S.J.; Jonsson, S.R.; Xiong, Y. Maedi-visna virus Vif protein uses motifs distinct from HIV-1 Vif to bind zinc and the cofactor required for A3 degradation. J. Biol. Chem. 2021, 296, 100045. [Google Scholar] [CrossRef]
- Du, J.; Rui, Y.; Zheng, W.; Li, P.; Kang, J.; Zhao, K.; Sun, T.; Yu, X.F. Vif-CBFβ interaction is essential for Vif-induced cell cycle arrest. Biochem. Biophys. Res. Commun. 2019, 511, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Magnusdottir, A.; Stenmark, P.; Flodin, S.; Nyman, T.; Kotenyova, T.; Graslund, S.; Ogg, D.; Nordlund, P. The structure of the PP2A regulatory subunit B56 gamma: The remaining piece of the PP2A jigsaw puzzle. Proteins 2009, 74, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Hertz, E.P.T.; Kruse, T.; Davey, N.E.; López-Méndez, B.; Sigurðsson, J.O.; Montoya, G.; Olsen, J.V.; Nilsson, J. A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol. Cell 2016, 63, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Garvanska, D.H.; Nasa, I.; Ueki, Y.; Zhang, G.; Kettenbach, A.N.; Peti, W.; Nilsson, J.; Page, R. A dynamic charge-charge interaction modulates PP2A:B56 substrate recruitment. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Parsons, G.G.; Spencer, C.A. Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. Mol. Cell. Biol. 1997, 17, 5791–5802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronja, I.; Orr-Weaver, T.L. Translational regulation of the cell cycle: When, where, how and why? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3638–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuer, R.; Becker, M.S.; Brechmann, M.; Mock, T.; Arnold, R.; Krammer, P.H. The protein phosphatase 2A regulatory subunit B56γ mediates suppression of T cell receptor (TCR)-induced nuclear factor-κB (NF-κB) activity. J. Biol. Chem. 2014, 289, 14996–15004. [Google Scholar] [CrossRef] [Green Version]
- Salamango, D.J.; Harris, R.S. Demystifying Cell Cycle Arrest by HIV-1 Vif. Trends Microbiol. 2021, 29, 381–384. [Google Scholar] [CrossRef]
- Fan, Y.; Sanyal, S.; Bruzzone, R. Breaking Bad: How Viruses Subvert the Cell Cycle. Front. Cell. Infect. Microbiol. 2018, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Elder, R.T. Viral infections and cell cycle G2/M regulation. Cell Res. 2005, 15, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.N. B′-protein phosphatase 2A is a functional binding partner of delta-retroviral integrase. Nucleic Acids Res. 2016, 44, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, V.; Shi, K.; Salamango, D.J.; Moeller, N.H.; Pandey, K.K.; Bera, S.; Aihara, H. Structural basis of host protein hijacking in human T-cell leukemia virus integration. Nat. Commun. 2020, 11, 3121. [Google Scholar] [CrossRef] [PubMed]
- Kruse, T.; Biedenkopf, N.; Hertz, E.P.T.; Dietzel, E.; Stalmann, G.; López-Méndez, B.; Davey, N.E.; Nilsson, J.; Becker, S. The Ebola Virus Nucleoprotein Recruits the Host PP2A-B56 Phosphatase to Activate Transcriptional Support Activity of VP30. Mol. Cell 2018, 69, 136–145.e6. [Google Scholar] [CrossRef] [PubMed]
- Roopchand, D.E.; Lee, J.M.; Shahinian, S.; Paquette, D.; Bussey, H.; Branton, P.E. Toxicity of human adenovirus E4orf4 protein in Saccharomyces cerevisiae results from interactions with the Cdc55 regulatory B subunit of PP2A. Oncogene 2001, 20, 5279–5290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dove, B.; Brooks, G.; Bicknell, K.; Wurm, T.; Hiscox, J.A. Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J. Virol. 2006, 80, 4147–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luperchio, A.M.; Jónsson, S.R.; Salamango, D.J. Evolutionary Conservation of PP2A Antagonism and G2/M Cell Cycle Arrest in Maedi-Visna Virus Vif. Viruses 2022, 14, 1701. https://doi.org/10.3390/v14081701
Luperchio AM, Jónsson SR, Salamango DJ. Evolutionary Conservation of PP2A Antagonism and G2/M Cell Cycle Arrest in Maedi-Visna Virus Vif. Viruses. 2022; 14(8):1701. https://doi.org/10.3390/v14081701
Chicago/Turabian StyleLuperchio, Adeline M., Stefán R. Jónsson, and Daniel J. Salamango. 2022. "Evolutionary Conservation of PP2A Antagonism and G2/M Cell Cycle Arrest in Maedi-Visna Virus Vif" Viruses 14, no. 8: 1701. https://doi.org/10.3390/v14081701
APA StyleLuperchio, A. M., Jónsson, S. R., & Salamango, D. J. (2022). Evolutionary Conservation of PP2A Antagonism and G2/M Cell Cycle Arrest in Maedi-Visna Virus Vif. Viruses, 14(8), 1701. https://doi.org/10.3390/v14081701