
Protein-Gene Orthology in Baculoviridae: An Exhaustive Analysis to Redefine the Ancestrally Common Coding Sequences
 , , , and
, , , and
Abstract
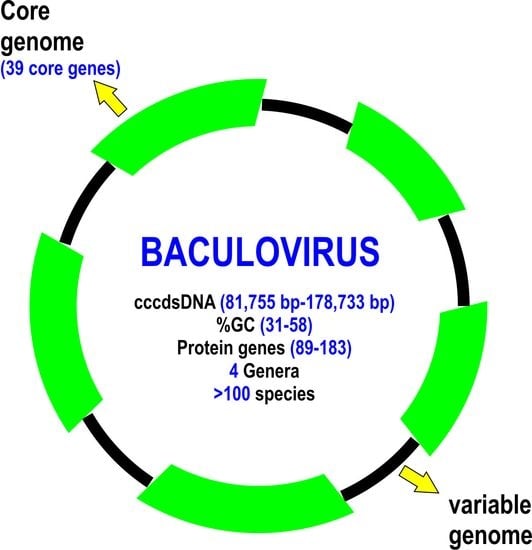
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Baculovirus Genome Data Set
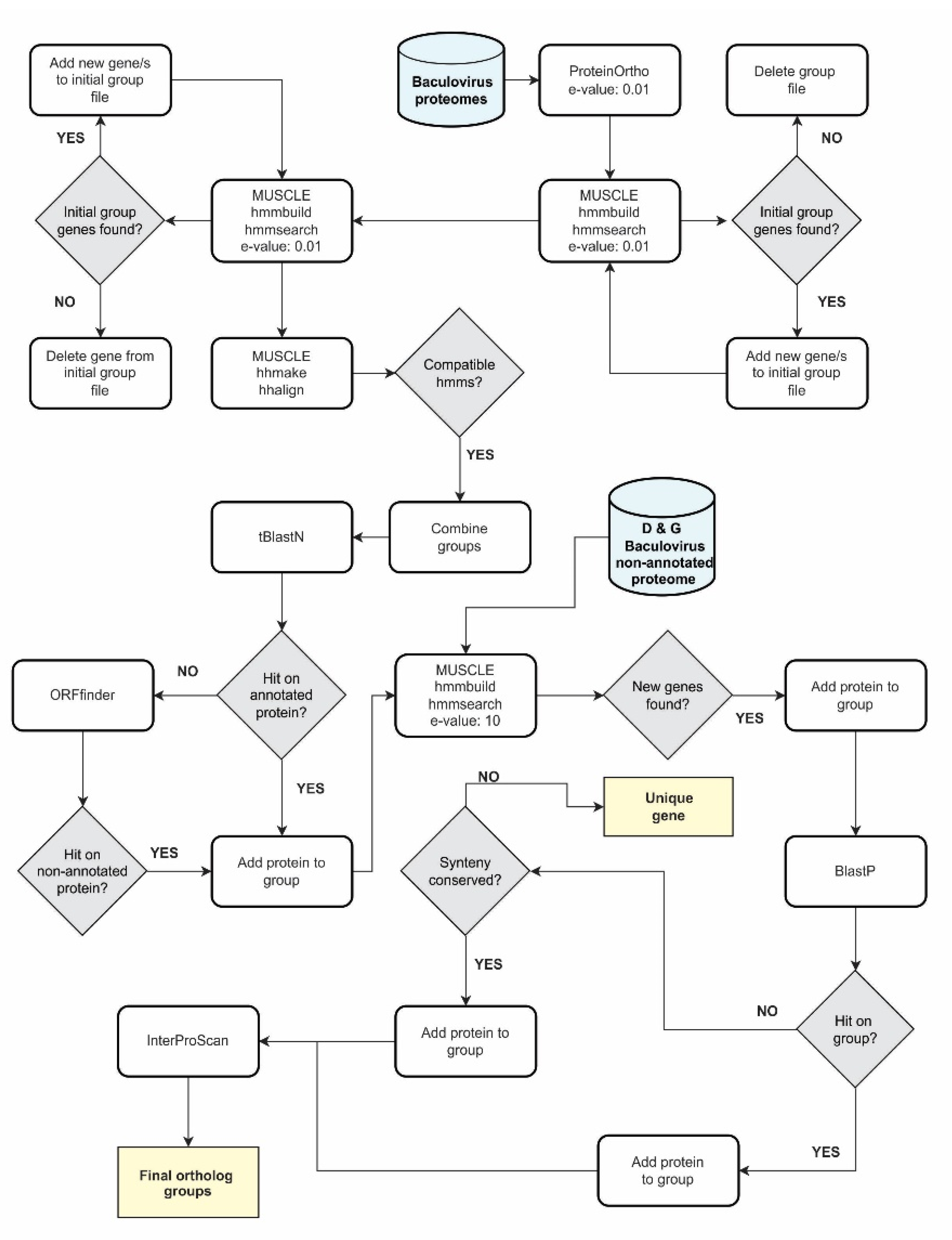
2.2. Ortholog Bioinformatic Pipeline for Baculovirus Genes
2.3. Orthologs Shared with Other Insect Viruses
2.4. Baculoviral Phylogeny Inference
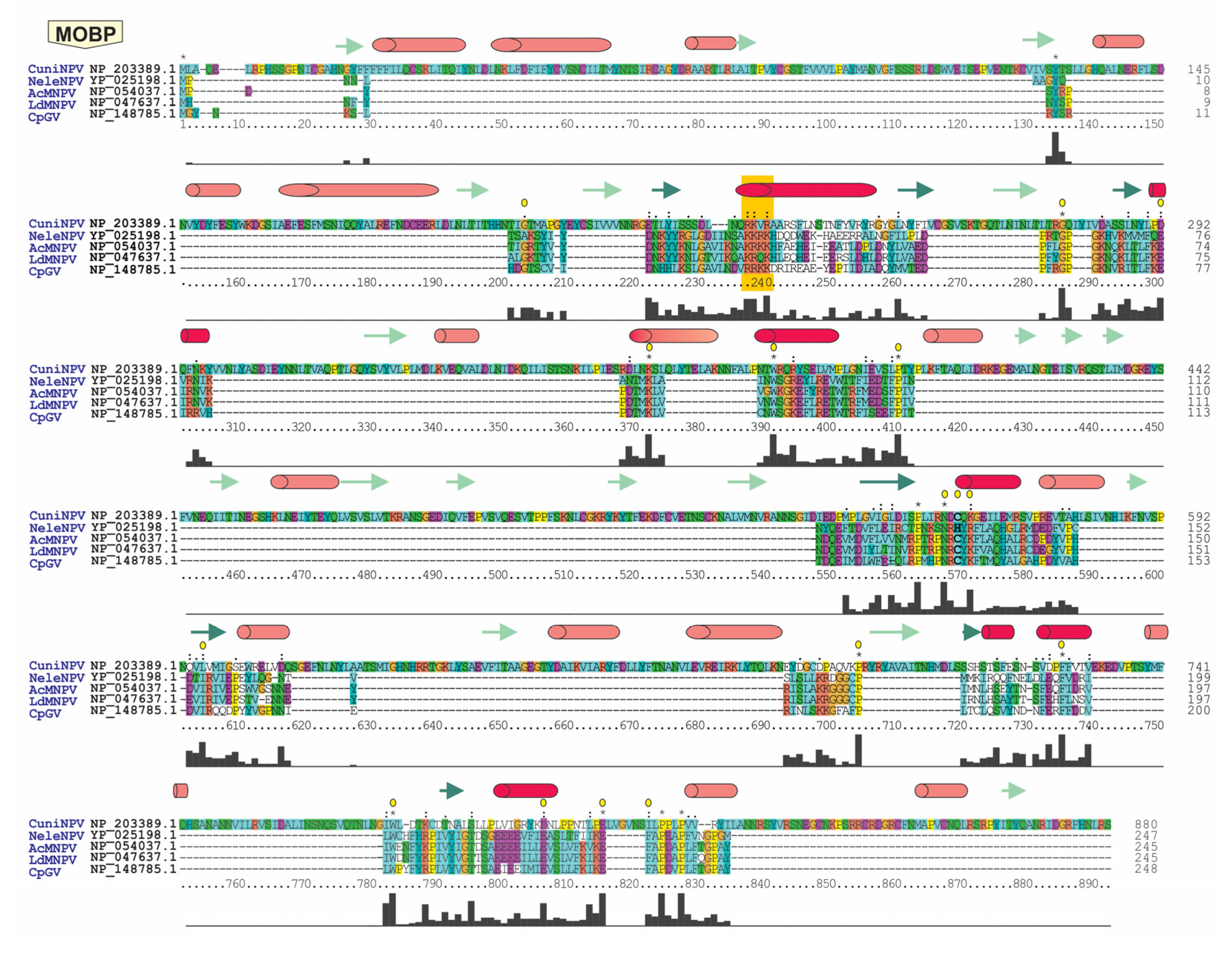
2.5. Protein Bioinformatic Studies
3. Results
3.1. Baculovirus Genomes
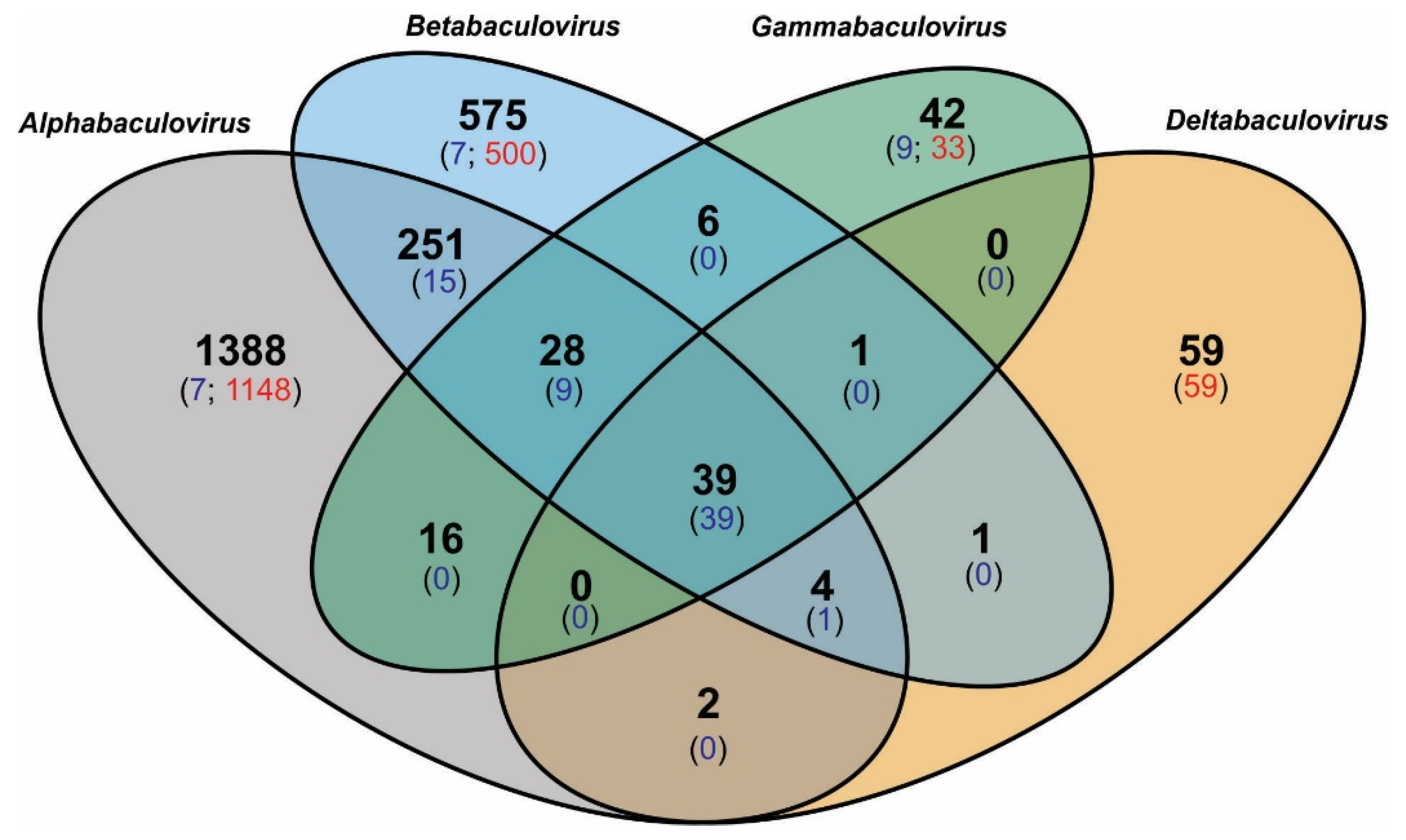
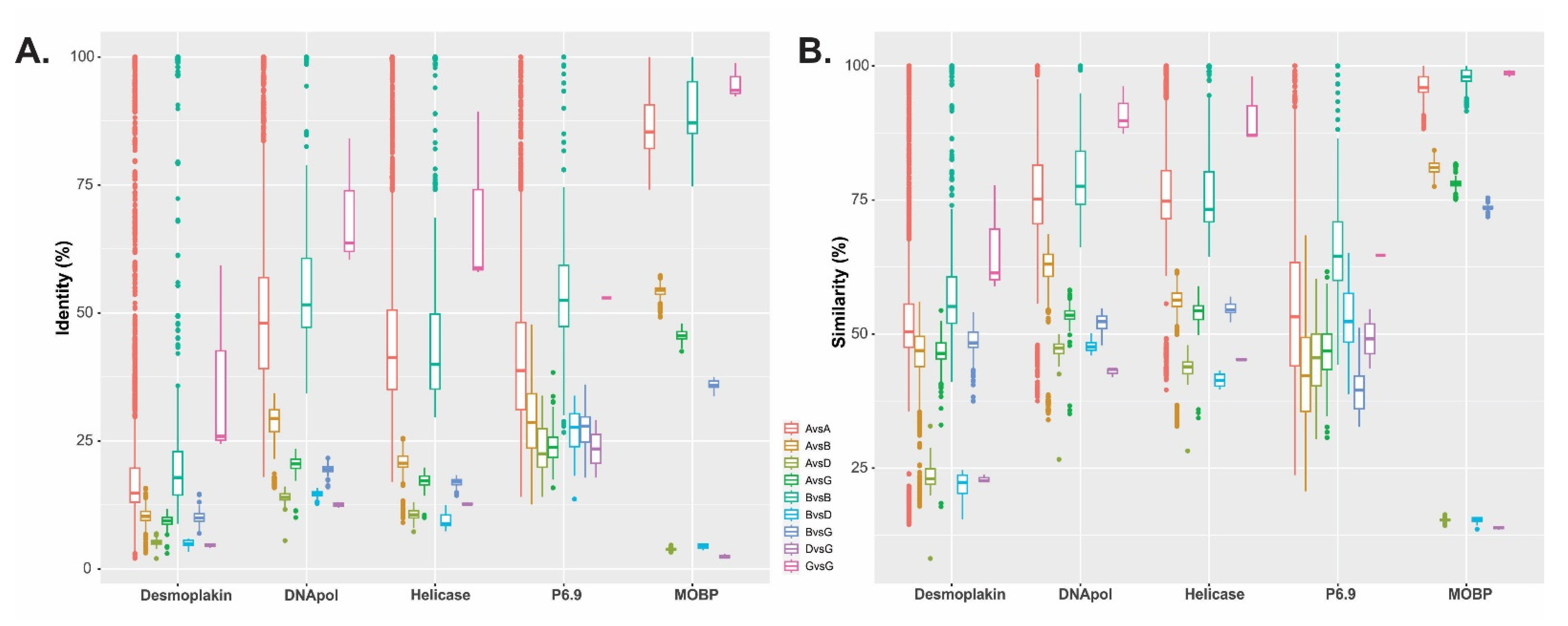
3.2. Baculovirus Shared and Unique Protein Genes
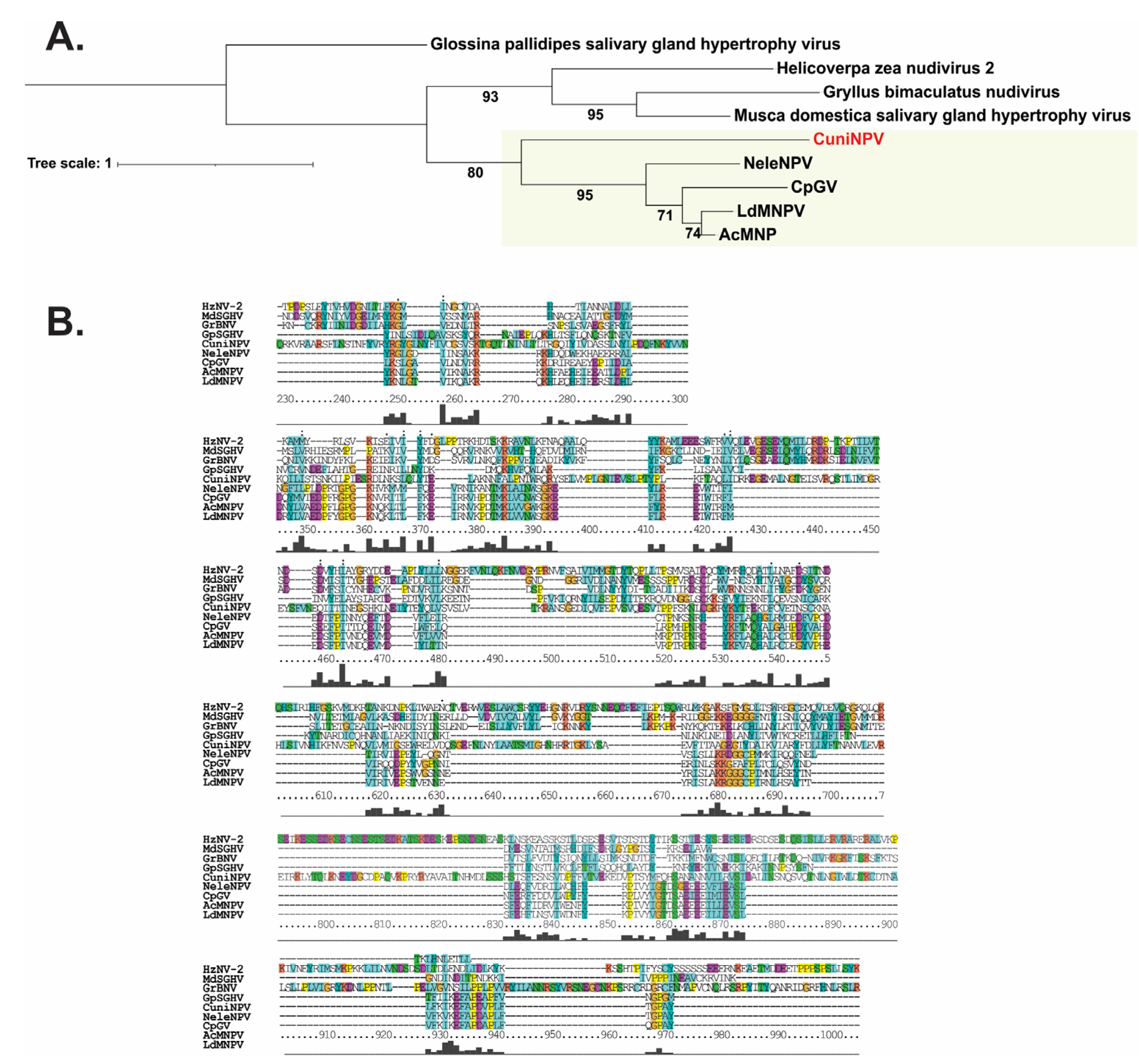
3.3. Baculovirus Phylogeny
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lacey, L.A.; Grzywacz, D.; Shapiro-Ilan, D.I.; Frutos, R.; Brownbridge, M.; Goettel, M.S. Insect pathogens as biological control agents: Back to the future. J. Invertebr. Pathol. 2015, 132, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Ono, C.; Okamoto, T.; Abe, T.; Matsuura, Y. Baculovirus as a Tool for Gene Delivery and Gene Therapy. Viruses 2018, 10, 510. [Google Scholar] [CrossRef]
- Targovnik, A.M.; Simonin, J.A.; Mc Callum, G.J.; Smith, I.; Cuccovia Warlet, F.U.; Nugnes, M.V.; Miranda, M.V.; Belaich, M.N. Solutions against emerging infectious and noninfectious human diseases through the application of baculovirus technologies. Appl. Microbiol. Biotechnol. 2021, 105, 8195–8226. [Google Scholar] [CrossRef]
- Rohrmann, G.F. Baculovirus Molecular Biology, 4th ed.; NCBI: Bethesda, MD, USA, 2019.
- Harrison, R.L.; Herniou, E.A.; Jehle, J.A.; Theilmann, D.A.; Burand, J.P.; Becnel, J.J.; Krell, P.J.; van Oers, M.M.; Mowery, J.D.; Bauchan, G.R. ICTV Virus Taxonomy Profile: Baculoviridae. J. Gen. Virol. 2018, 99, 1185–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hu, Z. Advances in Molecular Biology of Baculoviruses. Curr. Issues Mol. Biol. 2020, 34, 183–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shang, Y.; Chen, C.; Liu, S.; Chang, M.; Zhang, N.; Hu, H.; Zhang, F.; Zhang, T.; Wang, Z.; et al. Baculovirus Per Os Infectivity Factor Complex: Components and Assembly. J. Virol. 2019, 93, e02053-18. [Google Scholar] [CrossRef] [PubMed]
- Monsma, S.A.; Oomens, A.G.; Blissard, G.W. The GP64 envelope fusion protein is an essential baculovirus protein required for cell-to-cell transmission of infection. J. Virol. 1996, 70, 4607–4616. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, 161–164. [Google Scholar] [CrossRef]
- Jehle, J.A.; Blissard, G.W.; Bonning, B.C.; Cory, J.S.; Herniou, E.A.; Rohrmann, G.F.; Theilmann, D.A.; Thiem, S.M.; Vlak, J.M. On the classification and nomenclature of baculoviruses: A proposal for revision. Arch. Virol. 2006, 151, 1257–1266. [Google Scholar] [CrossRef]
- Lauzon, H.A.M.; Garcia-Maruniak, A.; de A Zanotto, P.M.; Clemente, J.C.; Herniou, E.A.; Lucarotti, C.J.; Arif, B.M.; Maruniak, J.E. Genomic comparison of Neodiprion sertifer and Neodiprion lecontei nucleopolyhedroviruses and identification of potential hymenopteran baculovirus-specific open reading frames. J. Gen. Virol. 2006, 87, 1477–1489. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.P.; Young, A.M.; Morin, B.; Lucarotti, C.J.; Koop, B.F.; Levin, D.B. Sequence analysis and organization of the Neodiprion abietis nucleopolyhedrovirus genome. J. Gen. Virol. 2007, 88, 1945–1951. [Google Scholar] [CrossRef]
- Afonso, C.L.; Tulman, E.R.; Lu, Z.; Balinsky, C.A.; Moser, B.A.; Becnel, J.J.; Rock, D.L.; Kutish, G.F. Genome sequence of a baculovirus pathogenic for Culex nigripalpus. J. Virol. 2001, 75, 11157–11165. [Google Scholar] [CrossRef] [PubMed]
- Miele, S.A.; Garavaglia, M.J.; Belaich, M.N.; Ghiringhelli, P.D. Baculovirus: Molecular insights on their diversity and conservation. Int. J. Evol. Biol. 2011, 2011, 379424. [Google Scholar] [CrossRef]
- Wang, J.; Xing, K.; Xiong, P.; Liang, H.; Zhu, M.; Zhao, J.; Yu, X.; Ning, X.; Li, R.; Wang, X. Identification of miRNAs encoded by Autographa californica nucleopolyhedrovirus. J. Gen. Virol. 2021, 102, 001510. [Google Scholar] [CrossRef] [PubMed]
- Wennmann, J.T.; Keilwagen, J.; Jehle, J.A. Baculovirus Kimura two-parameter species demarcation criterion is confirmed by the distances of 38 core gene nucleotide sequences. J. Gen. Virol. 2018, 99, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Garavaglia, M.J.; Miele, S.A.; Iserte, J.A.; Belaich, M.N.; Ghiringhelli, P.D. The ac53, ac78, ac101, and ac103 genes are newly discovered core genes in the family Baculoviridae. J. Virol. 2012, 86, 12069–12079. [Google Scholar] [CrossRef] [PubMed]
- Javed, M.A.; Biswas, S.; Willis, L.G.; Harris, S.; Pritchard, C.; van Oers, M.M.; Donly, B.C.; Erlandson, M.A.; Hegedus, D.D.; Theilmann, D.A. Autographa californica Multiple Nucleopolyhedrovirus AC83 is a Per Os Infectivity Factor (PIF) Protein Required for Occlusion-Derived Virus (ODV) and Budded Virus Nucleocapsid Assembly as well as Assembly of the PIF Complex in ODV Envelopes. J. Virol. 2017, 91, e02115-16. [Google Scholar] [CrossRef]
- Barrera, G.P.; Belaich, M.N.; Patarroyo, M.A.; Villamizar, L.F.; Ghiringhelli, P.D. Evidence of recent interspecies horizontal gene transfer regarding nucleopolyhedrovirus infection of Spodoptera frugiperda. BMC Genom. 2015, 16, 1008. [Google Scholar] [CrossRef]
- Nugnes, M.V.; Targovnik, A.M.; Mengual-Martí, A.; Miranda, M.V.; Cerrudo, C.S.; Herrero, S.; Belaich, M.N. The Membrane-Anchoring Region of the AcMNPV P74 Protein Is Expendable or Interchangeable with Homologs from Other Species. Viruses 2021, 13, 2416. [Google Scholar] [CrossRef] [PubMed]
- Gabaldón, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2013, 14, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Altenhoff, A.M.; Glover, N.M.; Dessimoz, C. Inferring Orthology and Paralogy. Methods. Mol. Biol. 2019, 1910, 149–175. [Google Scholar]
- Barrera, G.P.; Villamizar, L.F.; Araque, G.A.; Gómez, J.A.; Guevara, E.J.; Cerrudo, C.S.; Belaich, M.N. Natural Coinfection between Novel Species of Baculoviruses in Spodoptera ornithogalli Larvae. Viruses 2021, 13, 2520. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 25 January 2022).
- Van Rossum, G.; Drake, F.L., Jr. Python Tutorial; Centrum v oor Wiskunde en Informatica Amsterdam: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat. Commun. 2022, 13, 6968. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2022, 51, 418–427. [Google Scholar] [CrossRef]
- Minh, B.Q.; Hahn, M.W.; Lanfear, R. New Methods to Calculate Concordance Factors for Phylogenomic Datasets. Mol. Biol. Evol. 2020, 37, 2727–2733. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Lange, M.; Wang, H.; Hu, Z.; Wang, Y.; Hauschild, R. Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virology 2006, 346, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Bernhofer, M.; Dallago, C.; Karl, T.; Satagopam, V.; Heinzinger, M.; Littmann, M.; Olenyi, T.; Qiu, J.; Schütze, K.; Yachdav, G.; et al. PredictProtein—Predicting Protein Structure and Function for 29 Years. Nucleic Acids Res. 2021, 49, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Lipman, D.; Pearson, W. Rapid and sensitive protein similarity searches. Science 1985, 227, 1435–1444. [Google Scholar] [CrossRef]
- Peris, G.; Marzal, A. Normalized global alignment for pro-tein sequences. J. Theor. Biol. 2011, 291, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P. The Sequence Manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Perera, O.P.; Valles, S.M.; Green, T.B.; White, S.; Strong, C.A.; Becnel, J.J. Molecular analysis of an occlusion body protein from Culex nigripalpus nucleopolyhedrovirus (CuniNPV). J. Invertebr. Pathol. 2006, 91, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, F.; Chiu, E.; Gutmann, S.; Rajendran, C.; Haebel, P.W.; Ikeda, K.; Mori, H.; Ward, V.K.; Schulze-Briese, C.; Metcalf, P. The atomic structure of baculovirus polyhedra reveals the independent emergence of infectious crystals in DNA and RNA viruses. Proc. Natl. Acad. Sci. USA 2009, 106, 22205–22210. [Google Scholar] [CrossRef]
- Ji, X.; Sutton, G.; Evans, G.; Axford, D.; Owen, R.; Stuart, D.I. How baculovirus polyhedra fit square pegs into round holes to robustly package viruses. EMBO J. 2010, 29, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Mikhailovsky, G.E.; Gordon, R. LUCA to LECA, the Lucacene: A model for the gigayear delay from the first prokaryote to eukaryogenesis. Biosystems 2021, 205, 104415. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479–480, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.L.; Herniou, E.A.; Jehle, J.A.; Theilmann, D.A.; Burand, J.P.; Krell, P.J.; van Oers, M.M.; Vlak, J.M. Create Two New Genera and Eight New Species (Lefavirales: Nudiviridae). 2020. Available online: https://ictv.global/ictv/proposals/2020.005D.R.Nudiviridae_2ngen_8nsp.zip (accessed on 20 March 2023).
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, M.; Wang, X.; Liu, B. A comprehensive review and comparison of different computational methods for protein remote homology detection. Brief. Bioinform. 2018, 19, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhao, S.; Chen, N.; Wu, X. Molecular mechanism responsible for the hyperexpression of baculovirus polyhedrin. Gene 2022, 814, 146129. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G.F. Polyhedrin structure. Gen Virol. 1986, 67, 1499–1513. [Google Scholar] [CrossRef] [PubMed]
- Summers, M.D.; Smith, G.E. Comparative studies of baculovirus granulins and polyhedrins. Intervirology 1975, 6, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Maruniak, A.; Maruniak, J.E.; Zanotto, P.M.; Doumbouya, A.E.; Liu, J.C.; Merritt, T.M.; Lanoie, J.S. Sequence analysis of the genome of the Neodiprion sertifer nucleopolyhedrovirus. J. Virol. 2004, 78, 7036–7071. [Google Scholar] [CrossRef]
- Lauzon, H.A.; Lucarotti, C.J.; Krell, P.J.; Feng, Q.; Retnakaran, A.; Arif, B.M. Sequence and organization of the Neodiprion lecontei nucleopolyhedrovirus genome. J. Virol. 2004, 78, 7023–7035. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bininda-Emonds, O.R.; Van Oers, M.M.; Vlak, J.M.; Jehle, J.A. The genome of Oryctes rhinoceros nudivirus provides novel insight into the evolution of nuclear arthropod-specific large circular double-stranded DNA viruses. Virus Genes 2011, 42, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Bézier, A.; Thézé, J.; Gavory, F.; Gaillard, J.; Poulain, J.; Drezen, J.M.; Herniou, E.A. The genome of the nucleopolyhedrosis-causing virus from Tipula oleracea sheds new light on the Nudiviridae family. J. Virol. 2015, 89, 3008–3025. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Abd-Alla, A.M.; Wang, Y. Phylogeny and evolution of Hytrosaviridae. J. Invertebr. Pathol. 2013, 112, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, K.; Fu, Y. Ac106/107 affects production of infectious progeny BV by regulating transcription of late viral genes and host cell energy metabolism. Pest. Manag. Sci. 2021, 77, 4758–4769. [Google Scholar] [CrossRef]
- Dong, Z.Q.; Hu, N.; Dong, F.F.; Chen, T.T.; Jiang, Y.M.; Chen, P.; Lu, C.; Pan, M.H. Baculovirus LEF-11 Hijack Host ATPase ATAD3A to Promote Virus Multiplication in Bombyx mori cells. Sci. Rep. 2017, 7, 46187. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhang, X.; Xiao, M.; Li, K.; Wang, J.; Chen, P.; Hu, Z.; Lu, C.; Pan, M. Baculovirus LEF-11 interacts with BmIMPI to induce cell cycle arrest in the G2/M phase for viral replication. Pestic. Biochem. Physiol. 2022, 188, 105231. [Google Scholar] [CrossRef]
- Drezen, J.M.; Gauthier, J.; Josse, T.; Bézier, A.; Herniou, E.; Huguet, E. Foreign DNA acquisition by invertebrate genomes. J. Invertebr. Pathol. 2017, 147, 157–168. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerrudo, C.S.; Motta, L.F.; Cuccovia Warlet, F.U.; Lassalle, F.M.; Simonin, J.A.; Belaich, M.N. Protein-Gene Orthology in Baculoviridae: An Exhaustive Analysis to Redefine the Ancestrally Common Coding Sequences. Viruses 2023, 15, 1091. https://doi.org/10.3390/v15051091
Cerrudo CS, Motta LF, Cuccovia Warlet FU, Lassalle FM, Simonin JA, Belaich MN. Protein-Gene Orthology in Baculoviridae: An Exhaustive Analysis to Redefine the Ancestrally Common Coding Sequences. Viruses. 2023; 15(5):1091. https://doi.org/10.3390/v15051091
Chicago/Turabian StyleCerrudo, Carolina Susana, Lucas Federico Motta, Franco Uriel Cuccovia Warlet, Fernando Maku Lassalle, Jorge Alejandro Simonin, and Mariano Nicolás Belaich. 2023. "Protein-Gene Orthology in Baculoviridae: An Exhaustive Analysis to Redefine the Ancestrally Common Coding Sequences" Viruses 15, no. 5: 1091. https://doi.org/10.3390/v15051091