
Recent Clinical Isolates of Enterovirus D68 Have Increased Replication and Induce Enhanced Epithelial Immune Response Compared to the Prototype Fermon Strain
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
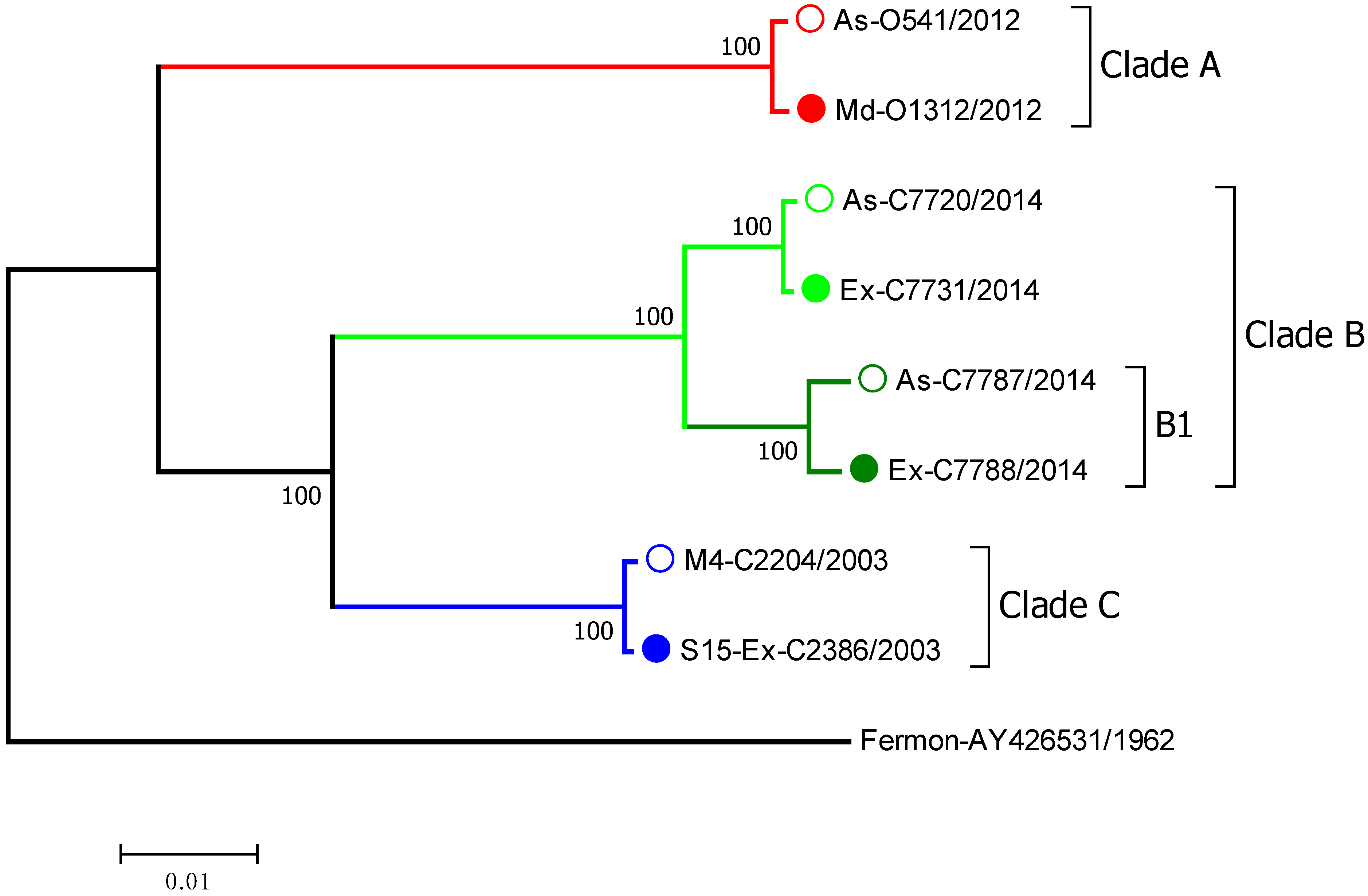
2.1. Virus Isolate Selection
2.2. Construction of the Full-Length EV-D68 cDNA Infectious Clones
2.3. Recombinant Virus Production in WisL Cells
2.4. Virus Purification
2.5. Reverse Transcription
2.6. Quantitative PCR
2.7. Differentiated BEC Cultures Grown at an Air–Liquid Interface
2.8. Infection of BEC-ALI Cultures
2.9. HeLa Cell Infections
2.10. RNA-Sequencing Library Construction and Sequencing of Directional Libraries
2.11. Statistical Analysis
3. Results
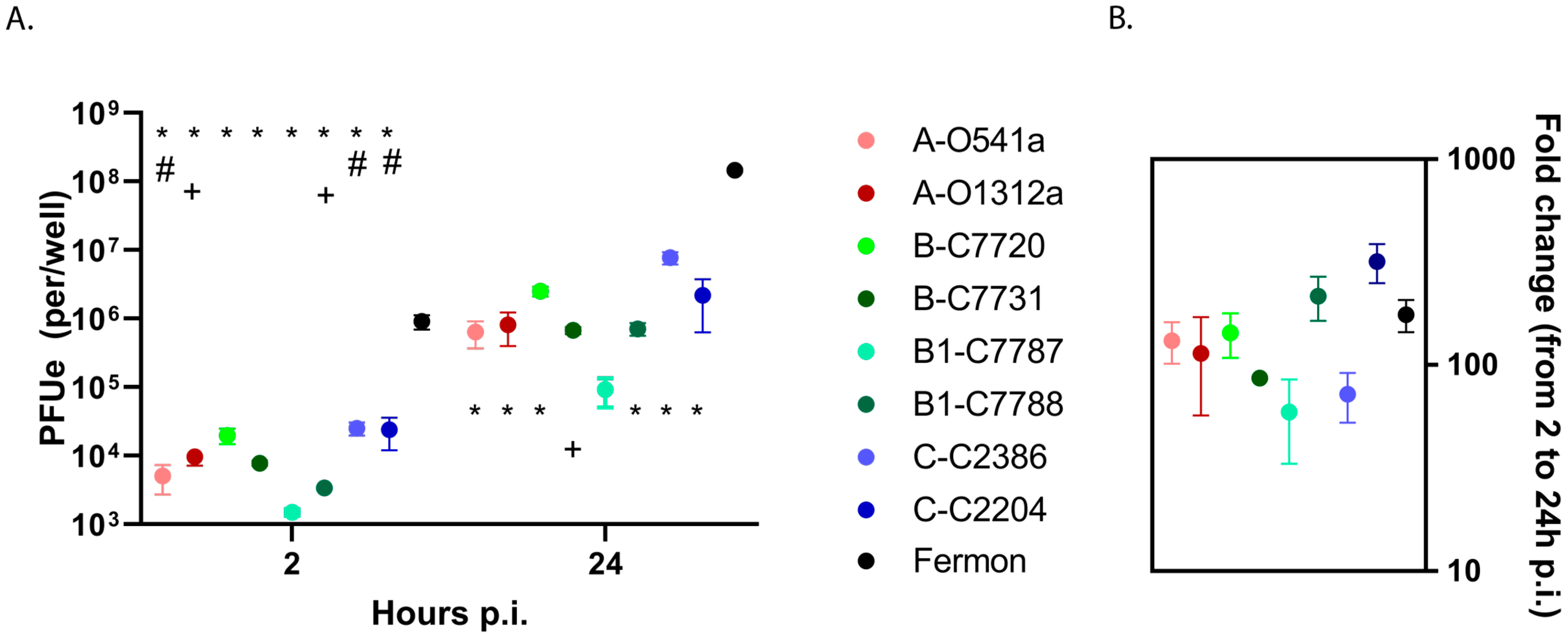
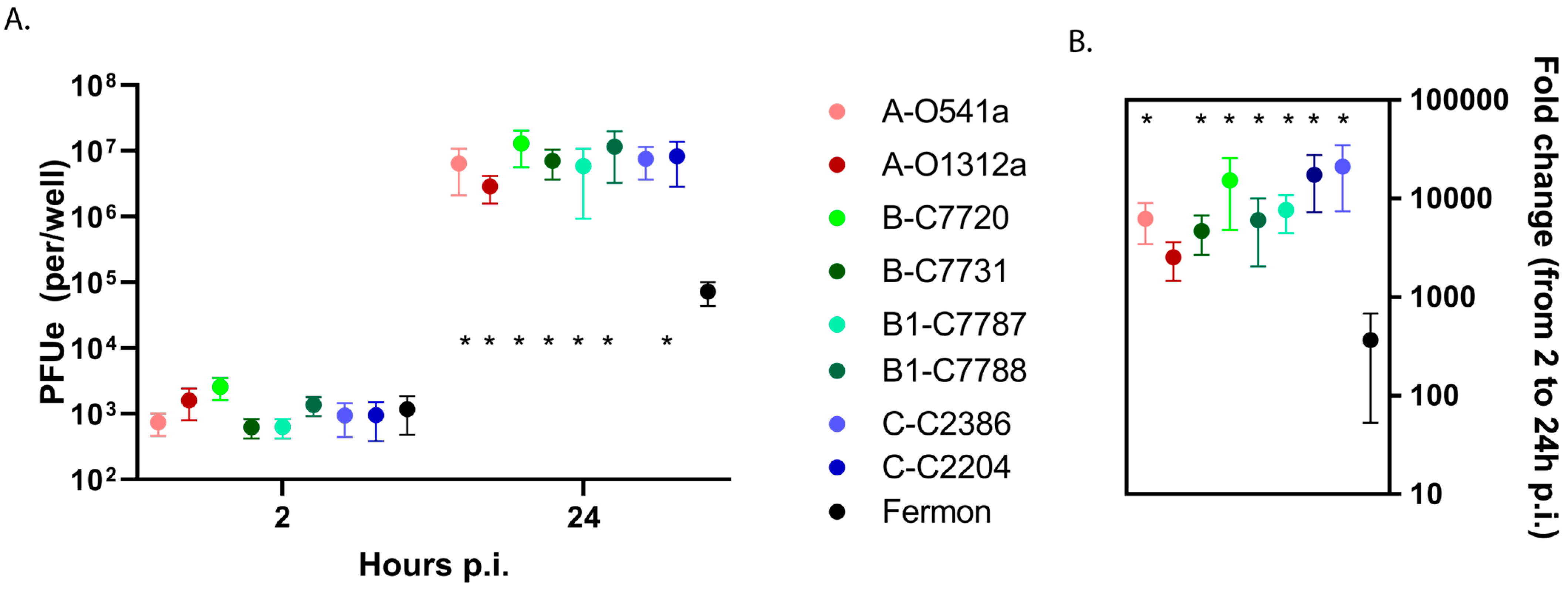
3.1. EV-D68 Binding and Replication in HeLa Cells
3.2. Time Course of EV-D68 Binding and Replication in Primary BECs
3.3. EV-D68 Binding and Replication in Primary BEC-ALI Cultures
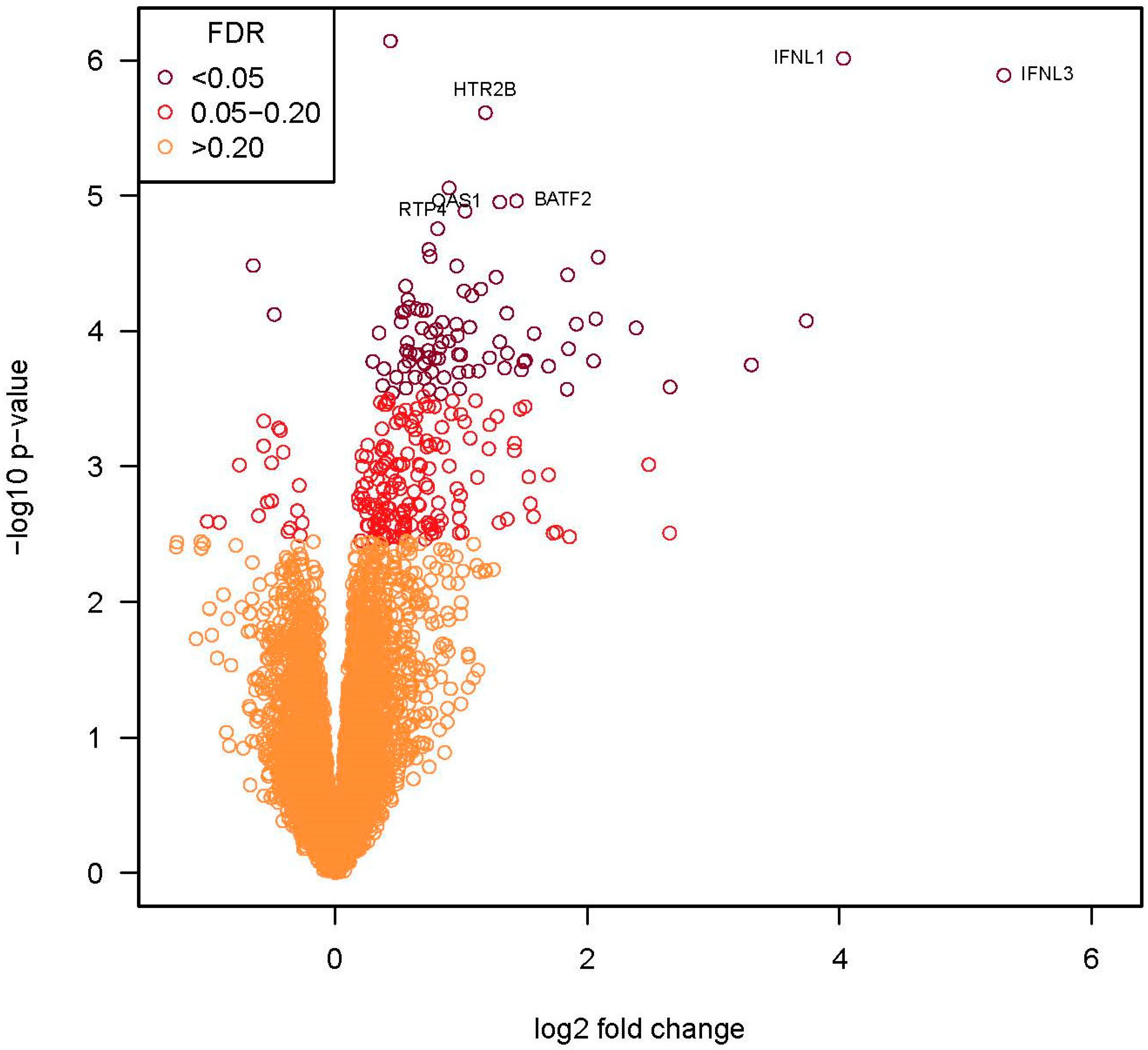
3.4. Assessment of Host Immune Response to EV-D68 by RNA-Seq Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schieble, J.H.; Fox, V.L.; Lennette, E.H. A probable new human picornavirus associated with respiratory diseases. Am. J. Epidemiol. 1967, 85, 297–310. [Google Scholar] [CrossRef]
- Greninger, A.L.; Naccache, S.N.; Messacar, K.; Clayton, A.; Yu, G.; Somasekar, S.; Federman, S.; Stryke, D.; Anderson, C.; Yagi, S.; et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the usa (2012–14): A retrospective cohort study. Lancet Infect. Dis. 2015, 15, 671–682. [Google Scholar] [CrossRef]
- Meijer, A.; van der Sanden, S.; Snijders, B.E.; Jaramillo-Gutierrez, G.; Bont, L.; van der Ent, C.K.; Overduin, P.; Jenny, S.L.; Jusic, E.; van der Avoort, H.G.; et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in the netherlands in 2010. Virology 2012, 423, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Suzuki, A.; Lupisan, S.; Okamoto, M.; Aniceto, R.; Egos, R.J.; Daya, E.E.; Tamaki, R.; Saito, M.; Fuji, N.; et al. Molecular evolution of enterovirus 68 detected in the philippines. PLoS ONE 2013, 8, e74221. [Google Scholar] [CrossRef] [PubMed]
- Oermann, C.M.; Schuster, J.E.; Conners, G.P.; Newland, J.G.; Selvarangan, R.; Jackson, M.A. Enterovirus D68. A focused review and clinical highlights from the 2014 U.S. Outbreak. Ann. Am. Thorac. Soc. 2015, 12, 775–781. [Google Scholar] [CrossRef]
- Hamparian, V.V.; Colonno, R.J.; Cooney, M.K.; Dick, E.C.; Gwaltney, J.M., Jr.; Hughes, J.H.; Jordan, W.S., Jr.; Kapikian, A.Z.; Mogabgab, W.J.; Monto, A.; et al. A collaborative report: Rhinoviruses--extension of the numbering system from 89 to 100. Virology 1987, 159, 191–192. [Google Scholar]
- Ishiko, H.; Miura, R.; Shimada, Y.; Hayashi, A.; Nakajima, H.; Yamazaki, S.; Takeda, N. Human rhinovirus 87 identified as human enterovirus 68 by vp4-based molecular diagnosis. Intervirology 2002, 45, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Okamoto, M.; Nakakita, S.; Suzuki, A.; Saito, M.; Tamaki, R.; Lupisan, S.; Roy, C.N.; Hiramatsu, H.; Sugawara, K.E.; et al. Antigenic and receptor binding properties of enterovirus 68. J. Virol. 2014, 88, 2374–2384. [Google Scholar] [CrossRef]
- Wei, W.; Guo, H.; Chang, J.; Yu, Y.; Liu, G.; Zhang, N.; Willard, S.H.; Zheng, S.; Yu, X.F. ICAM-5/telencephalin is a functional entry receptor for enterovirus D68. Cell Host Microbe 2016, 20, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Baggen, J.; Liu, Y.; Lyoo, H.; van Vliet, A.L.W.; Wahedi, M.; de Bruin, J.W.; Roberts, R.W.; Overduin, P.; Meijer, A.; Rossmann, M.G.; et al. Bypassing pan-enterovirus host factor pla2g16. Nat. Commun. 2019, 10, 3171. [Google Scholar] [CrossRef]
- Rosenfeld, A.B.; Warren, A.L.; Racaniello, V.R. Neurotropism of enterovirus D68 isolates is independent of sialic acid and is not a recently acquired phenotype. mBio 2019, 10, e02370-19. [Google Scholar] [CrossRef]
- Khetsuriani, N.; Lamonte-Fowlkes, A.; Oberst, S.; Pallansch, M.A. Enterovirus surveillance--united states, 1970–2005. MMWR Surveill. Summ. 2006, 55, 1–20. [Google Scholar] [PubMed]
- Tokarz, R.; Firth, C.; Madhi, S.A.; Howie, S.R.; Wu, W.; Sall, A.A.; Haq, S.; Briese, T.; Lipkin, W.I. Worldwide emergence of multiple clades of enterovirus 68. J. Gen. Virol. 2012, 93, 1952–1958. [Google Scholar] [CrossRef]
- Midgley, S.E.; Benschop, K.; Dyrdak, R.; Mirand, A.; Bailly, J.L.; Bierbaum, S.; Buderus, S.; Böttcher, S.; Eis-Hübinger, A.M.; Hönemann, M.; et al. Co-circulation of multiple enterovirus D68 subclades, including a novel b3 cluster, across europe in a season of expected low prevalence, 2019/20. Eurosurveillance 2020, 25, 1900749. [Google Scholar] [CrossRef]
- Xiang, Z.; Li, L.; Lei, X.; Zhou, H.; Zhou, Z.; He, B.; Wang, J. Enterovirus 68 3c protease cleaves trif to attenuate antiviral responses mediated by toll-like receptor 3. J. Virol. 2014, 88, 6650–6659. [Google Scholar] [CrossRef]
- Lemanske, R.F. The childhood origins of asthma (coast) study. Pediatr. Allergy Immunol. 2002, 13 (Suppl. S15), 38–43. [Google Scholar] [CrossRef]
- Teach, S.J.; Gill, M.A.; Togias, A.; Sorkness, C.A.; Arbes, S.J.; Calatroni, A.; Wildfire, J.J.; Gergen, P.J.; Cohen, R.T.; Pongracic, J.A.; et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J. Allergy Clin. Immunol. 2015, 136, 1476–1485. [Google Scholar] [CrossRef]
- Gern, J.E.; Visness, C.M.; Gergen, P.J.; Wood, R.A.; Bloomberg, G.R.; O’Connor, G.T.; Kattan, M.; Sampson, H.A.; Witter, F.R.; Sandel, M.T.; et al. The urban environment and childhood asthma (ureca) birth cohort study: Design, methods, and study population. BMC Pulm. Med. 2009, 9, 17. [Google Scholar] [CrossRef]
- Huang, W.; Wang, G.; Zhuge, J.; Nolan, S.M.; Dimitrova, N.; Fallon, J.T. Whole-genome sequence analysis reveals the enterovirus D68 isolates during the united states 2014 outbreak mainly belong to a novel clade. Sci. Rep. 2015, 5, 15223. [Google Scholar] [CrossRef]
- Herold, J.; Andino, R. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. J. Virol. 2000, 74, 6394–6400. [Google Scholar] [CrossRef]
- Lee, W.M.; Wang, W. Human rhinovirus type 16: Mutant v1210a requires capsid-binding drug for assembly of pentamers to form virions during morphogenesis. J. Virol. 2003, 77, 6235–6244. [Google Scholar] [CrossRef]
- Nakagome, K.; Bochkov, Y.A.; Ashraf, S.; Brockman-Schneider, R.A.; Evans, M.D.; Pasic, T.R.; Gern, J.E. Effects of rhinovirus species on viral replication and cytokine production. J. Allergy Clin. Immunol. 2014, 134, 332–341. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. The r package rsubread is easier, faster, cheaper and better for alignment and quantification of rna sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Carlson, M. Org.Hs.Eg.Db: Genome Wide Annotation for Human. R package 3.3.0. 2019. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html (accessed on 22 May 2023).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Freeman, M.C.; Wells, A.I.; Ciomperlik-Patton, J.; Myerburg, M.M.; Yang, L.; Konopka-Anstadt, J.; Coyne, C.B. Respiratory and intestinal epithelial cells exhibit differential susceptibility and innate immune responses to contemporary EV-D68 isolates. eLife 2021, 10, e66687. [Google Scholar] [CrossRef]
- Baggen, J.; Thibaut, H.J.; Staring, J.; Jae, L.T.; Liu, Y.; Guo, H.; Slager, J.J.; de Bruin, J.W.; van Vliet, A.L.; Blomen, V.A.; et al. Enterovirus d68 receptor requirements unveiled by haploid genetics. Proc. Natl. Acad. Sci. USA 2016, 113, 1399–1404. [Google Scholar] [CrossRef]
- Sridhar, A.; Depla, J.A.; Mulder, L.A.; Karelehto, E.; Brouwer, L.; Kruiswijk, L.; Vieira de Sá, R.; Meijer, A.; Evers, M.M.; van Kuppeveld, F.J.M.; et al. Enterovirus D68 infection in human primary airway and brain organoids: No additional role for heparan sulfate binding for neurotropism. Microbiol. Spectr. 2022, 10, e0169422. [Google Scholar] [CrossRef]
- Blomqvist, S.; Savolainen, C.; Raman, L.; Roivainen, M.; Hovi, T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J. Clin. Microbiol. 2002, 40, 4218–4223. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. Biovenn—A web application for the comparison and visualization of biological lists using area-proportional venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Strain | GenBank Accession Number | Restriction Enzyme (3′ End) | Restriction Enzyme (5′ End) |
|---|---|---|---|
| Lab Fermon | AY426531 | Sall | XhoI |
| O541a | KX255369 | MluI | Sall |
| O1312a | KX255358 | MluI | SaII |
| C2386 | KX255388 | PstI | XhoI |
| C2204 | KX255410 | PstI | XhoI |
| C7720 | KX255352 | BstEII | XhoI |
| C7731 | KX255354 | BstEII | XhoI |
| C7787 | KX255382 | SpelI | XhoI |
| C7788 | KX255413 | SpelI | XhoI |
| Gene Name | Gene Symbol | Fold Change | p-Value | FDR p-Value |
|---|---|---|---|---|
| Interferon lambda 3 | IFNL3 | 39.41 | 1 × 10−6 | 7 × 10−3 |
| Interferon lambda 1 | IFNL1 | 16.33 | 1 × 10−6 | 7 × 10−3 |
| Interferon lambda 2 | IFNL2 | 13.32 | 8 × 10−5 | 4 × 10−2 |
| Interferon gamma-induced protein 10 | CXCL10 | 9.85 | 2 × 10−4 | 4 × 10−2 |
| Interferon gamma-induced protein 11 | CXCL11 | 6.30 | 3 × 10−4 | 5 × 10−2 |
| Interferon-induced protein tetratriopeptide repeats 2 | IFIT2 | 5.23 | 1 × 10−4 | 4 × 10−2 |
| 2′-5′-oligoadenylate synthase | OASL | 4.24 | 3 × 10−5 | 4 × 10−2 |
| Z-DNA-binding protein 1 | ZBP1 | 4.19 | 8 × 10−5 | 4 × 10−2 |
| Interferon-induced protein tetratriopeptide repeats 1 | IFIT1 | 4.14 | 2 × 10−4 | 4 × 10−2 |
| Interferon-induced protein tetratriopeptide repeats 3 | IFIT3 | 3.77 | 9 × 10−5 | 4 × 10−2 |
| Radical SAM domain-containing 2 | RSAD2 | 3.60 | 1 × 10−4 | 4 × 10−2 |
| Cytidine/uridine monophosphate kinase 2 | CMPK2 | 3.59 | 4 × 10−5 | 4 × 10−2 |
| Interferon-induced GTP-binding protein Mx2 | MX2 | 3.58 | 3 × 10−4 | 5 × 10−2 |
| Interferon-induced protein AIM2 | AIM2 | 3.23 | 2 × 10−4 | 4 × 10−2 |
| Tumor necrosis factor receptor superfamily member 13B | TNFSF13B | 2.98 | 1 × 10−4 | 4 × 10−2 |
| Probable E3 ubiquitin-protein ligase HERC5 | HERC5 | 2.85 | 2 × 10−4 | 4 × 10−2 |
| Interferon-induced GTP-binding protein MX1 | MX1 | 2.83 | 2 × 10−4 | 4 × 10−2 |
| Epithelial stromal interaction 1 | EPSTI1 | 2.78 | 2 × 10−4 | 4 × 10−2 |
| Basic leucine zipper transcription factor, ATF-like 2 | BATF2 | 2.71 | 1 × 10−5 | 2 × 10−2 |
| 2′-5′-oligoadenylate synthetase 2 | OAS2 | 2.58 | 1 × 10−4 | 4 × 10−2 |
| RIG-I (retinoic acid-inducible gene I) | DDX58 | 2.57 | 7 × 10−5 | 4 × 10−2 |
| 2′-5′-oligoadenylate synthetase 3 | OAS3 | 2.54 | 2 × 10−4 | 4 × 10−2 |
| 2′-5′-oligoadenylate synthetase 1 | OAS1 | 2.47 | 1 × 10−5 | 2 × 10−2 |
| Probable E3 ubiquitin-protein ligase HERC6 | HERC6 | 2.47 | 1 × 10−4 | 4 × 10−2 |
| Ubiquitin-specific peptidase 18 | USP18 | 2.42 | 4 × 10−5 | 4 × 10−2 |
| XIAP-associated factor 1 | XAF1 | 2.34 | 2 × 10−4 | 4 × 10−2 |
| 5-Hydroxytryptamine receptor 2B | HTR2B | 2.28 | 2 × 10−6 | 9 × 10−3 |
| T-cell activation RhoGTPase-activating protein | TAGAP | 2.23 | 5 × 10−5 | 4 × 10−2 |
| Interferon-induced transmembrane protein 1 | IFITM1 | 2.20 | 2 × 10−4 | 4 × 10−2 |
| Interferon-induced with helicase C domain 1 | IFIH1 | 2.12 | 6 × 10−5 | 4 × 10−2 |
| Interferon-induced guanylate-binding protein 1 | GBP4 | 2.10 | 9 × 10−5 | 4 × 10−2 |
| Probable ATP-dependent RNA helicase 60 | DDX60 | 2.08 | 2 × 10−4 | 4 × 10−2 |
| Receptor transporter protein 4 | RTP4 | 2.04 | 1 × 10−5 | 2 × 10−2 |
| Hematopoietic SH2 Domain | HSH2D | 2.03 | 5 × 10−5 | 4 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devries, M.K.; Bochkov, Y.A.; Evans, M.D.; Gern, J.E.; Jackson, D.J. Recent Clinical Isolates of Enterovirus D68 Have Increased Replication and Induce Enhanced Epithelial Immune Response Compared to the Prototype Fermon Strain. Viruses 2023, 15, 1291. https://doi.org/10.3390/v15061291
Devries MK, Bochkov YA, Evans MD, Gern JE, Jackson DJ. Recent Clinical Isolates of Enterovirus D68 Have Increased Replication and Induce Enhanced Epithelial Immune Response Compared to the Prototype Fermon Strain. Viruses. 2023; 15(6):1291. https://doi.org/10.3390/v15061291
Chicago/Turabian StyleDevries, Mark K., Yury A. Bochkov, Michael D. Evans, James E. Gern, and Daniel J. Jackson. 2023. "Recent Clinical Isolates of Enterovirus D68 Have Increased Replication and Induce Enhanced Epithelial Immune Response Compared to the Prototype Fermon Strain" Viruses 15, no. 6: 1291. https://doi.org/10.3390/v15061291