
Transcriptome Profiling of Vero E6 Cells during Original Parental or Cell-Attenuated Porcine Epidemic Diarrhea Virus Infection

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses, Cells, and Antibody
2.2. Indirect Immunofluorescence Assay (IFA)
2.3. RNA Extraction and Sequencing
2.4. Quantification of Gene Expression Level
2.5. Gene Function Enrichment Analysis
2.6. Additional Bioinformatic Methods
2.7. Real-Time Quantitative PCR
2.8. Western Blotting
2.9. Measurement of Cytokines in Cell Supernatants
3. Results
3.1. Establishment of Cellular Model for PEDV Infection
3.2. Assessment of RNA-Sequencing Quality
3.3. Weighted Gene Co-Expression Network Analysis (WGCNA)
3.4. Identification of Differentially Expressed Genes in PEDV-Infected Cells
3.5. Gene Ontology (GO) Enrichment Analysis of DEGs
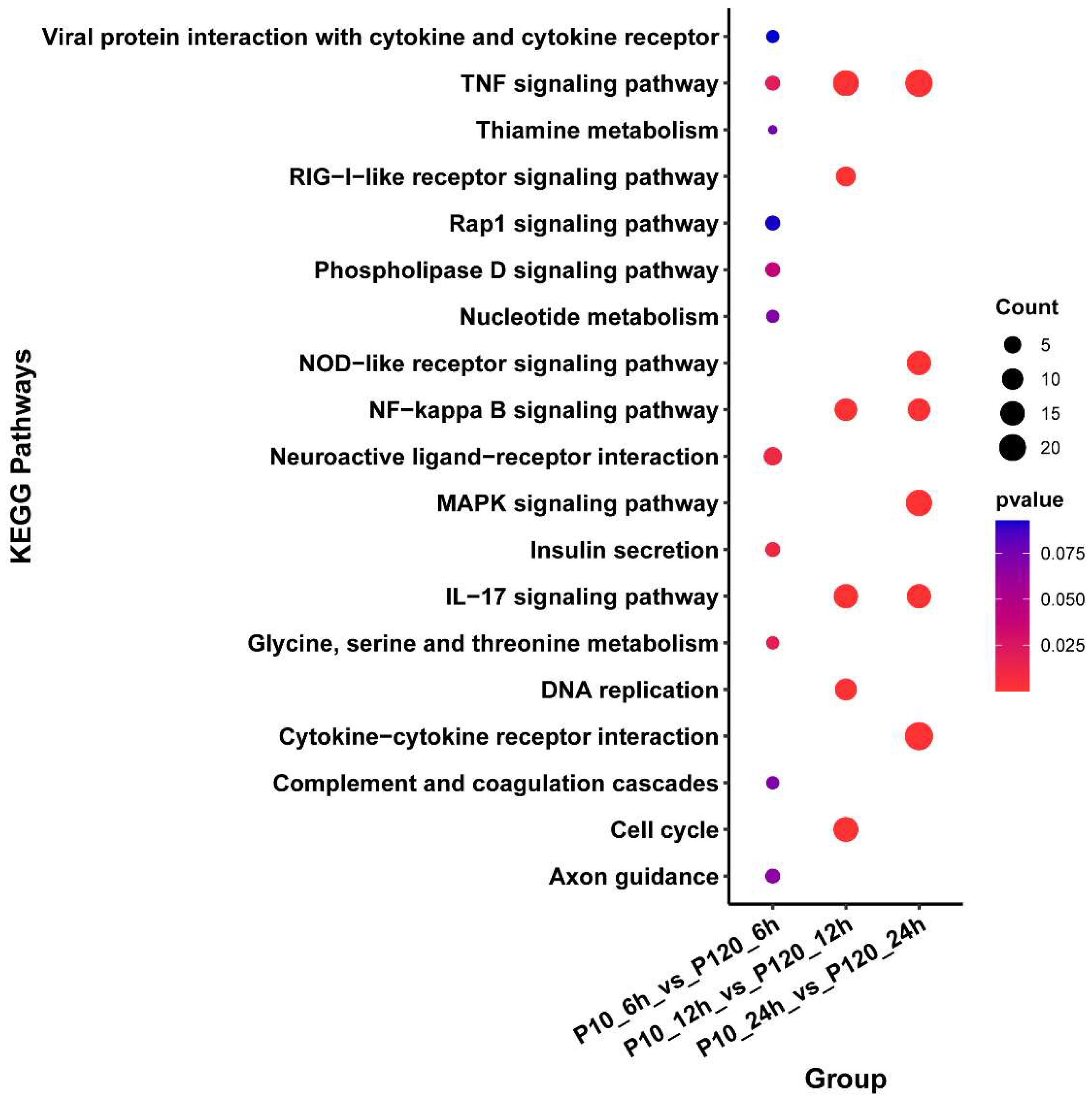
3.6. KEGG Enrichment Analysis of DEGs
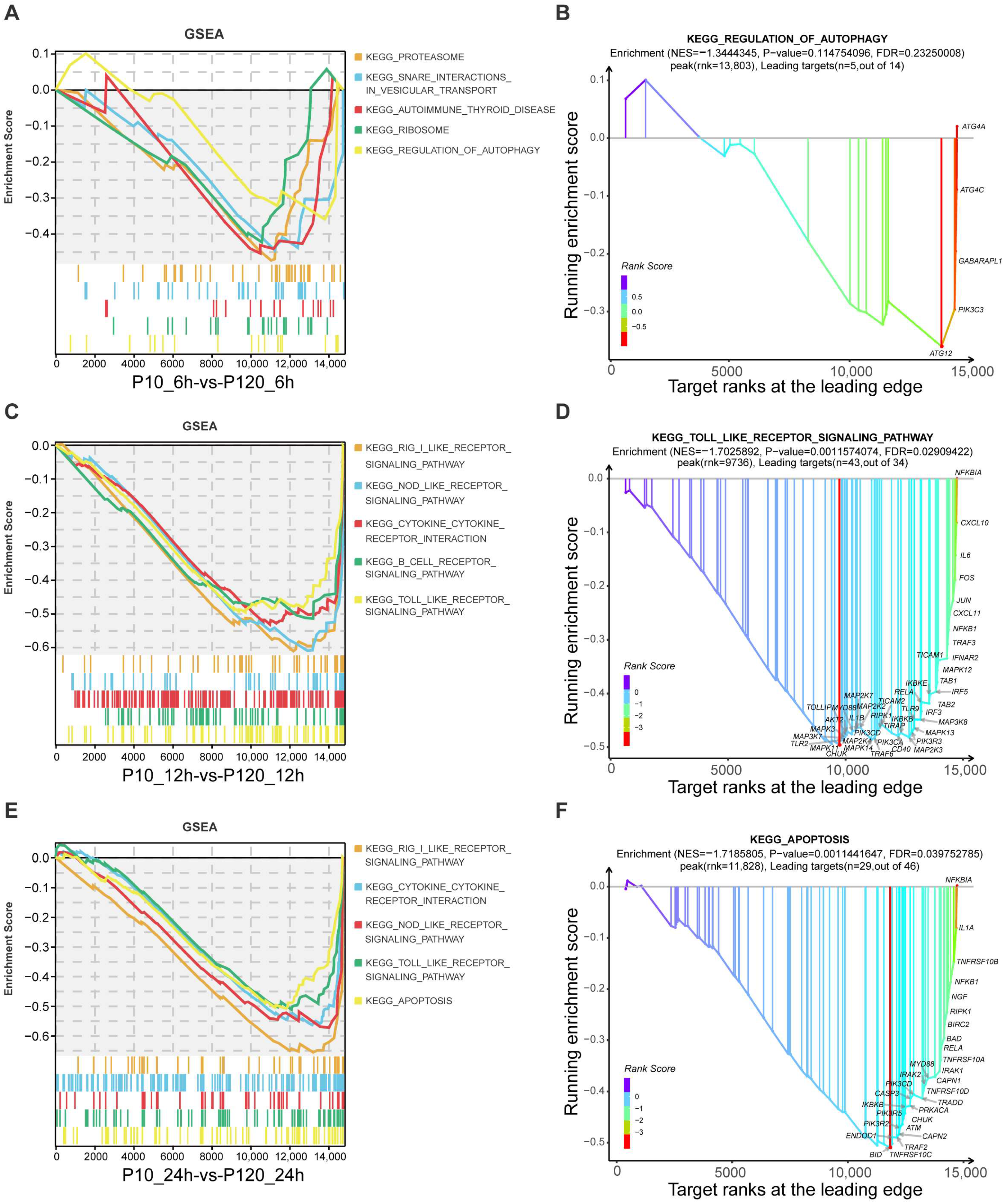
3.7. GSEA Pathways Enrichment Analysis
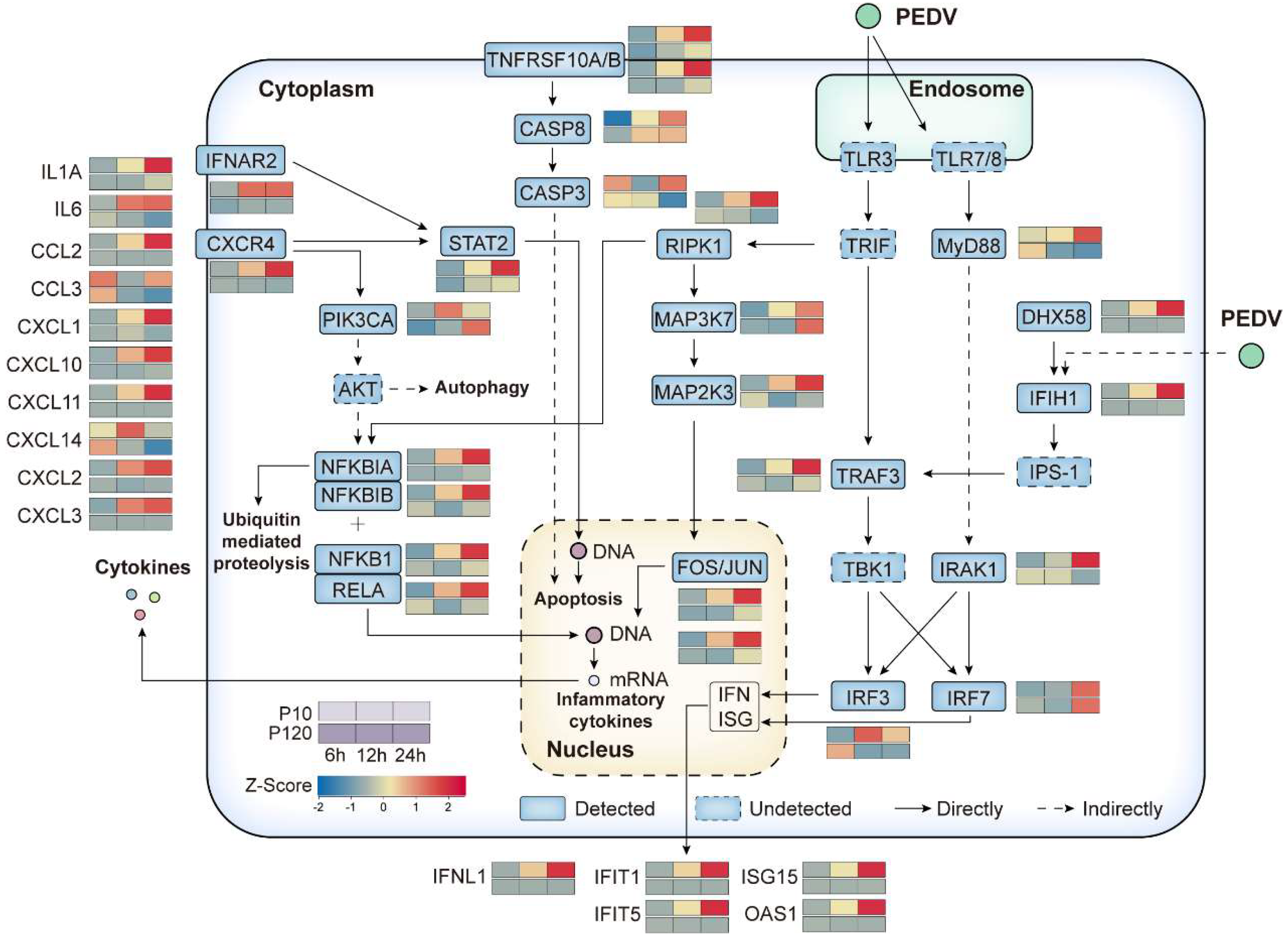
3.8. Immune-Related Pathways Involved in PEDV Infection
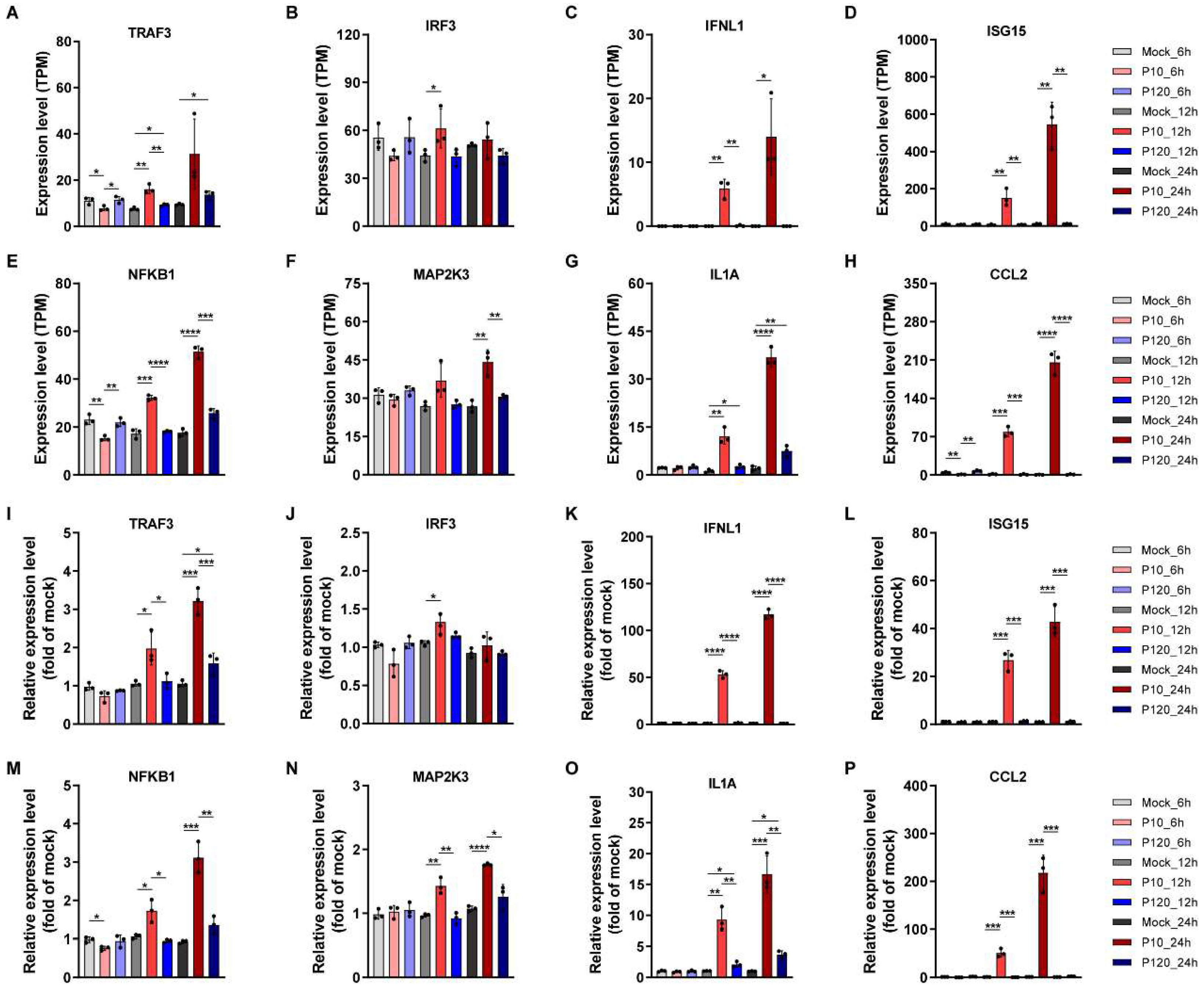
3.9. Validation of RNA-Seq Data by Quantitative Real-Time PCR
3.10. Validation of RNA-Seq Data by Western Blotting or ELISA
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jung, K.; Saif, L.J.; Wang, Q. Porcine epidemic diarrhea virus (pedv): An update on etiology, transmission, pathogenesis, and prevention and control. Virus Res. 2020, 286, 198045. [Google Scholar]
- Li, W.; Li, H.; Liu, Y.; Pan, Y.; Deng, F.; Song, Y.; Tang, X.; He, Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012, 18, 1350–1353. [Google Scholar]
- Lee, C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol. J. 2015, 12, 193. [Google Scholar]
- Hou, Y.; Wang, Q. Emerging highly virulent porcine epidemic diarrhea virus: Molecular mechanisms of attenuation and rational design of live attenuated vaccines. Int. J. Mol. Sci. 2019, 20, 5478. [Google Scholar]
- Chasey, D.; Cartwright, S.F. Virus-like particles associated with porcine epidemic diarrhoea. Res. Vet. Sci. 1978, 25, 255–256. [Google Scholar] [CrossRef]
- Lin, C.M.; Saif, L.J.; Marthaler, D.; Wang, Q. Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Res. 2016, 226, 20–39. [Google Scholar]
- Wang, L.; Byrum, B.; Zhang, Y. New variant of porcine epidemic diarrhea virus, united states, 2014. Emerg. Infect. Dis. 2014, 20, 917–919. [Google Scholar]
- Sun, Y.; Chen, Y.; Han, X.; Yu, Z.; Wei, Y.; Zhang, G. Porcine epidemic diarrhea virus in Asia: An alarming threat to the global pig industry. Infect. Genet. Evol. 2019, 70, 24–26. [Google Scholar]
- Li, C.; Lu, H.; Geng, C.; Yang, K.; Liu, W.; Liu, Z.; Yuan, F.; Gao, T.; Wang, S.; Wen, P.; et al. Epidemic and evolutionary characteristics of swine enteric viruses in south-central China from 2018 to 2021. Viruses 2022, 14, 1420. [Google Scholar]
- Niu, X.; Wang, Q. Prevention and control of porcine epidemic diarrhea: The development of recombination-resistant live attenuated vaccines. Viruses 2022, 14, 1317. [Google Scholar]
- Gao, Q.; Zheng, Z.; Wang, H.; Yi, S.; Zhang, G.; Gong, L. The new porcine epidemic diarrhea virus outbreak may mean that existing commercial vaccines are not enough to fully protect against the epidemic strains. Front. Vet. Sci. 2021, 8, 697839. [Google Scholar]
- Wu, Y.; Li, W.; Zhou, Q.; Li, Q.; Xu, Z.; Shen, H.; Chen, F. Characterization and pathogenicity of vero cell-attenuated porcine epidemic diarrhea virus ct strain. Virol. J. 2019, 16, 121. [Google Scholar]
- Mutz, K.O.; Heilkenbrinker, A.; Lonne, M.; Walter, J.G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar]
- Sun, J.; Ye, F.; Wu, A.; Yang, R.; Pan, M.; Sheng, J.; Zhu, W.; Mao, L.; Wang, M.; Xia, Z.; et al. Comparative transcriptome analysis reveals the intensive early stage responses of host cells to SARS-CoV-2 infection. Front. Microbiol. 2020, 11, 593857. [Google Scholar]
- Salgado-Albarran, M.; Navarro-Delgado, E.I.; Del, M.A.; Alcaraz, N.; Baumbach, J.; Gonzalez-Barrios, R.; Soto-Reyes, E. Comparative transcriptome analysis reveals key epigenetic targets in SARS-CoV-2 infection. NPJ Syst. Biol. Appl. 2021, 7, 21. [Google Scholar]
- Nardacci, R.; Colavita, F.; Castilletti, C.; Lapa, D.; Matusali, G.; Meschi, S.; Del, N.F.; Colombo, D.; Capobianchi, M.R.; Zumla, A.; et al. Evidences for lipid involvement in SARS-CoV-2 cytopathogenesis. Cell Death Dis. 2021, 12, 263. [Google Scholar]
- Yang, L.; Wang, C.; Shu, J.; Feng, H.; He, Y.; Chen, J.; Shu, J. Porcine epidemic diarrhea virus induces vero cell apoptosis via the p53-puma signaling pathway. Viruses 2021, 13, 1218. [Google Scholar]
- Barrett, P.N.; Mundt, W.; Kistner, O.; Howard, M.K. Vero cell platform in vaccine production: Moving towards cell culture-based viral vaccines. Expert Rev. Vaccines 2009, 8, 607–618. [Google Scholar]
- Xu, Z.; Lin, Y.; Zou, C.; Peng, P.; Wu, Y.; Wei, Y.; Liu, Y.; Gong, L.; Cao, Y.; Xue, C. Attenuation and characterization of porcine enteric alphacoronavirus strain gds04 via serial cell passage. Vet. Microbiol. 2019, 239, 108489. [Google Scholar]
- Park, J.E.; Kang, K.J.; Ryu, J.H.; Park, J.Y.; Jang, H.; Sung, D.J.; Kang, J.G.; Shin, H.J. Porcine epidemic diarrhea vaccine evaluation using a newly isolated strain from Korea. Vet. Microbiol. 2018, 221, 19–26. [Google Scholar]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincrnas. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by rna-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. Venndiagram: A package for the generation of highly-customizable venn and euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro ugene: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neubock, R.; Hofacker, I.L. The vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar]
- Langfelder, P.; Horvath, S. Wgcna: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar]
- Xu, Q.; Tang, Y.; Huang, G. Innate immune responses in RNA viral infection. Front. Med. 2021, 15, 333–346. [Google Scholar]
- Li, S.; Yang, J.; Zhu, Z.; Zheng, H. Porcine epidemic diarrhea virus and the host innate immune response. Pathogens 2020, 9, 367. [Google Scholar]
- Huang, Y.W.; Dickerman, A.W.; Pineyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the united states. MBio 2013, 4, e713–e737. [Google Scholar]
- Li, Y.; Wang, G.; Wang, J.; Man, K.; Yang, Q. Cell attenuated porcine epidemic diarrhea virus strain zhejiang08 provides effective immune protection attributed to dendritic cell stimulation. Vaccine 2017, 35, 7033–7041. [Google Scholar]
- Mick, E.; Kamm, J.; Pisco, A.O.; Ratnasiri, K.; Babik, J.M.; Castaneda, G.; Derisi, J.L.; Detweiler, A.M.; Hao, S.L.; Kangelaris, K.N.; et al. Upper airway gene expression reveals suppressed immune responses to SARS-CoV-2 compared with other respiratory viruses. Nat. Commun. 2020, 11, 5854. [Google Scholar]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.; Hosmillo, M.; Fossati, A.; et al. Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar]
- Peng, O.; Wei, X.; Ashraf, U.; Hu, F.; Xia, Y.; Xu, Q.; Hu, G.; Xue, C.; Cao, Y.; Zhang, H. Genome-wide transcriptome analysis of porcine epidemic diarrhea virus virulent or avirulent strain-infected porcine small intestinal epithelial cells. Virol. Sin. 2022, 37, 70–81. [Google Scholar]
- Mao, J.; Huang, X.; Shan, Y.; Xu, J.; Gao, Q.; Xu, X.; Zhang, C.; Shi, F.; Yue, M.; He, F.; et al. Transcriptome analysis revealed inhibition of lipid metabolism in 2-d porcine enteroids by infection with porcine epidemic diarrhea virus. Vet. Microbiol. 2022, 273, 109525. [Google Scholar]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation d614g alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar]
- Wei, D.; Yu, D.M.; Wang, M.J.; Zhang, D.H.; Cheng, Q.J.; Qu, J.M.; Zhang, X.X. Genome-wide characterization of the seasonal h3n2 virus in shanghai reveals natural temperature-sensitive strains conferred by the i668v mutation in the pa subunit. Emerg. Microbes Infect. 2018, 7, 171. [Google Scholar]
- Witteveldt, J.; Blundell, R.; Maarleveld, J.J.; Mcfadden, N.; Evans, D.J.; Simmonds, P. The influence of viral RNA secondary structure on interactions with innate host cell defences. Nucleic Acids Res. 2014, 42, 3314–3329. [Google Scholar]
- Hosseini, R.S.A.; Mclellan, A.D. Implications of SARS-CoV-2 mutations for genomic RNA structure and host microrna targeting. Int. J. Mol. Sci. 2020, 21, 4807. [Google Scholar]
- Hyde, J.L.; Gardner, C.L.; Kimura, T.; White, J.P.; Liu, G.; Trobaugh, D.W.; Huang, C.; Tonelli, M.; Paessler, S.; Takeda, K.; et al. A viral RNA structural element alters host recognition of nonself RNA. Science 2014, 343, 783–787. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence | Product Length (bp) |
|---|---|---|---|
| TRAF3 | Forward | 5′-CCTTGTTCCGATTTGGAGGTG-3′ | 300 |
| Reverse | 5′-TGACCCGGCTCCATTCTGTG-3′ | ||
| IRF3 | Forward | 5′-TTGTGACCTCAGGAGTTGGG-3′ | 249 |
| Reverse | 5′-GCTTCAGTGGGTTTTCACGG-3′ | ||
| IFNL1 | Forward | 5′-CGGGAATTGGGACCTAAGGC-3′ | 274 |
| Reverse | 5′-GCCAGGGGACTCCTTTTCGG-3′ | ||
| ISG15 | Forward | 5′-CACGGCCATGGGTAGGGA-3′ | 266 |
| Reverse | 5′-TCCTCACCAGGATGCTCAGT-3′ | ||
| NFKB1 | Forward | 5′-GGCTACCCTGGCACAGAAAT-3′ | 291 |
| Reverse | 5′-TCATCCCGGAGCTCGTCTAT-3′ | ||
| MAP2K3 | Forward | 5′-CCATCGGAGACAGGAACTTTGA-3′ | 260 |
| Reverse | 5′-GACGTCCAAGTCCATGAGCA-3′ | ||
| IL1A | Forward | 5′-GCCCGCAATCAAAGCATCAT-3′ | 217 |
| Reverse | 5′-GTGTCTCAGGCAGCTCCTTC-3′ | ||
| CCL2 | Forward | 5′-GTGTCCTAAAGAAGCAGTGATCTTC-3′ | 198 |
| Reverse | 5′-TCTGAGGGTATTTAGGGCAAGT-3′ | ||
| GAPDH | Forward | 5′-CGGAGTGAACGGATTTGGC-3′ | 248 |
| Reverse | 5′-CACCCCATTTGATGTTGGCG-3′ |
| Module | Blue | Yellow | Brown | Turquoise | Green | Red |
|---|---|---|---|---|---|---|
| Gene number | 1154 | 923 | 926 | 1470 | 238 | 48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, O.; Wu, Y.; Hu, F.; Xia, Y.; Geng, R.; Huang, Y.; Zeng, S.; Hu, G.; Xue, C.; Zhang, H.; et al. Transcriptome Profiling of Vero E6 Cells during Original Parental or Cell-Attenuated Porcine Epidemic Diarrhea Virus Infection. Viruses 2023, 15, 1426. https://doi.org/10.3390/v15071426
Peng O, Wu Y, Hu F, Xia Y, Geng R, Huang Y, Zeng S, Hu G, Xue C, Zhang H, et al. Transcriptome Profiling of Vero E6 Cells during Original Parental or Cell-Attenuated Porcine Epidemic Diarrhea Virus Infection. Viruses. 2023; 15(7):1426. https://doi.org/10.3390/v15071426
Chicago/Turabian StylePeng, Ouyang, Yu Wu, Fangyu Hu, Yu Xia, Rui Geng, Yihui Huang, Siying Zeng, Guangli Hu, Chunyi Xue, Hao Zhang, and et al. 2023. "Transcriptome Profiling of Vero E6 Cells during Original Parental or Cell-Attenuated Porcine Epidemic Diarrhea Virus Infection" Viruses 15, no. 7: 1426. https://doi.org/10.3390/v15071426