

Transcriptional Landscapes of Herelleviridae Bacteriophages and Staphylococcus aureus during Phage Infection: An Overview


Abstract
:1. Introduction
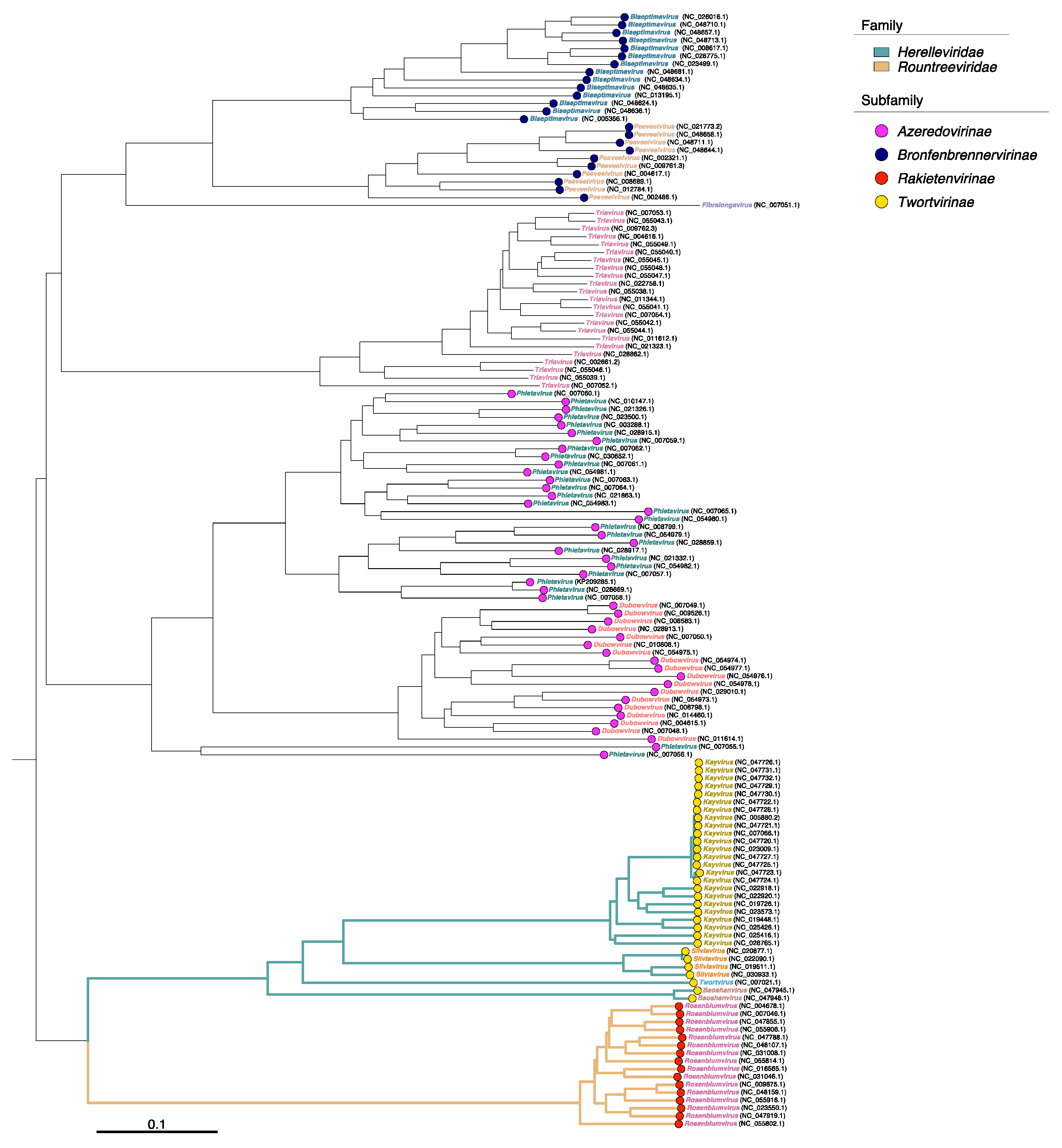
2. Virulent Staphylococcus aureus Phages
3. Features of the Transcriptomics Experiment for the Phage—Bacteria System
4. Herelleviridae Phage Gene Transcriptional Analysis
4.1. Analysis of Phage Promoters and Terminators
4.2. Transcriptional Characteristics of Herelleviridae Phages
5. Transcriptional Response of Staphylococcus aureus
6. Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salmond, G.P.C.; Fineran, P.C. A Century of the Phage: Past, Present and Future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Batinovic, S.; Wassef, F.; Knowler, S.A.; Rice, D.T.; Stanton, C.R.; Rose, J.; Franks, A.E. Bacteriophages in Natural and Artificial Environments. Pathogens 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danis-Wlodarczyk, K.M.; Wozniak, D.J.; Abedon, S.T. Treating Bacterial Infections with Bacteriophage-Based Enzybiotics: In Vitro, In Vivo and Clinical Application. Antibiotics 2021, 10, 1497. [Google Scholar] [CrossRef] [PubMed]
- Mendes, J.J.; Leandro, C.; Mottola, C.; Barbosa, R.; Silva, F.A.; Oliveira, M.; Vilela, C.L.; Melo-Cristino, J.; Górski, A.; Pimentel, M.; et al. In Vitro Design of a Novel Lytic Bacteriophage Cocktail with Therapeutic Potential against Organisms Causing Diabetic Foot Infections. J. Med. Microbiol. 2014, 63, 1055–1065. [Google Scholar] [CrossRef]
- Ooi, M.L.; Drilling, A.J.; Morales, S.; Fong, S.; Moraitis, S.; Macias-Valle, L.; Vreugde, S.; Psaltis, A.J.; Wormald, P.-J. Safety and Tolerability of Bacteriophage Therapy for Chronic Rhinosinusitis Due to Staphylococcus aureus. JAMA Otolaryngol. Neck Surg. 2019, 145, 723. [Google Scholar] [CrossRef] [PubMed]
- Petrovic Fabijan, A.; Lin, R.C.Y.; Ho, J.; Maddocks, S.; Ben Zakour, N.L.; Iredell, J.R.; Westmead Bacteriophage Therapy Team; Khalid, A.; Venturini, C.; Chard, R.; et al. Safety of Bacteriophage Therapy in Severe Staphylococcus Aureus Infection. Nat. Microbiol. 2020, 5, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus Aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [Green Version]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Li, Z.; Zhuang, H.; Wang, G.; Wang, H.; Dong, Y. Prevalence, Predictors, and Mortality of Bloodstream Infections Due to Methicillin-Resistant Staphylococcus Aureus in Patients with Malignancy: Systemic Review and Meta-Analysis. BMC Infect. Dis. 2021, 21, 74. [Google Scholar] [CrossRef]
- Siddiqui, A.H.; Koirala, J. Methicillin Resistant Staphylococcus Aureus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ferry, T.; Kolenda, C.; Batailler, C.; Gustave, C.-A.; Lustig, S.; Malatray, M.; Fevre, C.; Josse, J.; Petitjean, C.; Chidiac, C.; et al. Phage Therapy as Adjuvant to Conservative Surgery and Antibiotics to Salvage Patients With Relapsing S. Aureus Prosthetic Knee Infection. Front. Med. 2020, 7, 570572. [Google Scholar] [CrossRef]
- Doub, J.B.; Ng, V.Y.; Johnson, A.J.; Slomka, M.; Fackler, J.; Horne, B.; Brownstein, M.J.; Henry, M.; Malagon, F.; Biswas, B. Salvage Bacteriophage Therapy for a Chronic MRSA Prosthetic Joint Infection. Antibiotics 2020, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Sanchez, C.; Gonzales, F.; Buckley, M.; Biswas, B.; Henry, M.; Deschenes, M.V.; Horne, B.; Fackler, J.; Brownstein, M.J.; Schooley, R.T.; et al. Successful Treatment of Staphylococcus Aureus Prosthetic Joint Infection with Bacteriophage Therapy. Viruses 2021, 13, 1182. [Google Scholar] [CrossRef] [PubMed]
- Mulzer, J.; Trampuz, A.; Potapov, E.V. Treatment of Chronic Left Ventricular Assist Device Infection with Local Application of Bacteriophages. Eur. J. Cardiothorac. Surg. 2020, 57, 1003–1004. [Google Scholar] [CrossRef]
- Gupta, P.; Singh, H.S.; Shukla, V.K.; Nath, G.; Bhartiya, S.K. Bacteriophage Therapy of Chronic Nonhealing Wound: Clinical Study. Int. J. Low. Extrem. Wounds 2019, 18, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.; Kutter, E.; Bryan, D.; Wheat, G.; Kuhl, S. Resolving Digital Staphylococcal Osteomyelitis Using Bacteriophage—A Case Report. Antibiotics 2018, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.M.; Gaikwad, S.L.; Dhakephalkar, P.K.; Kothari, R.; Singh, R.P. Intriguing Interaction of Bacteriophage-Host Association: An Understanding in the Era of Omics. Front. Microbiol. 2017, 8, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutz, K.-O.; Heilkenbrinker, A.; Lönne, M.; Walter, J.-G.; Stahl, F. Transcriptome Analysis Using Next-Generation Sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef]
- Gondane, A.; Itkonen, H.M. Revealing the History and Mystery of RNA-Seq. Curr. Issues Mol. Biol. 2023, 45, 1860–1874. [Google Scholar] [CrossRef]
- Ceyssens, P.-J.; Minakhin, L.; Van Den Bossche, A.; Yakunina, M.; Klimuk, E.; Blasdel, B.; De Smet, J.; Noben, J.-P.; Bläsi, U.; Severinov, K.; et al. Development of Giant Bacteriophage ΦKZ Is Independent of the Host Transcription Apparatus. J. Virol. 2014, 88, 10501–10510. [Google Scholar] [CrossRef] [Green Version]
- Chevallereau, A.; Blasdel, B.G.; De Smet, J.; Monot, M.; Zimmermann, M.; Kogadeeva, M.; Sauer, U.; Jorth, P.; Whiteley, M.; Debarbieux, L.; et al. Next-Generation “-Omics” Approaches Reveal a Massive Alteration of Host RNA Metabolism during Bacteriophage Infection of Pseudomonas Aeruginosa. PLoS Genet. 2016, 12, e1006134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leskinen, K.; Blasdel, B.; Lavigne, R.; Skurnik, M. RNA-Sequencing Reveals the Progression of Phage-Host Interactions between ΦR1-37 and Yersinia Enterocolitica. Viruses 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojardín, L.; Salas, M. Global Transcriptional Analysis of Virus-Host Interactions between Phage Φ29 and Bacillus Subtilis. J. Virol. 2016, 90, 9293–9304. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Shen, M.; Jiang, X.; Shen, W.; Zhong, Q.; Yang, Y.; Tan, Y.; Agnello, M.; He, X.; Hu, F.; et al. Transcriptomic and Metabolomics Profiling of Phage–Host Interactions between Phage PaP1 and Pseudomonas Aeruginosa. Front. Microbiol. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacher, J.; Flint, A.; Butcher, J.; Blasdel, B.; Reynolds, H.; Lavigne, R.; Stintzi, A.; Szymanski, C. Transcriptomic Analysis of the Campylobacter Jejuni Response to T4-Like Phage NCTC 12673 Infection. Viruses 2018, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, B.W.; Logel, D.Y.; Mirzai, M.; Pascovici, D.; Molloy, M.P.; Jaschke, P.R. Proteomic and Transcriptomic Analysis of Microviridae ΦX174 Infection Reveals Broad Upregulation of Host Escherichia Coli Membrane Damage and Heat Shock Responses. mSystems 2021, 6, e00046-21. [Google Scholar] [CrossRef] [PubMed]
- Logel, D.Y.; Jaschke, P.R. A High-Resolution Map of Bacteriophage ΦX174 Transcription. Virology 2020, 547, 47–56. [Google Scholar] [CrossRef]
- Yang, Z.; Yin, S.; Li, G.; Wang, J.; Huang, G.; Jiang, B.; You, B.; Gong, Y.; Zhang, C.; Luo, X.; et al. Global Transcriptomic Analysis of the Interactions between Phage ΦAbp1 and Extensively Drug-Resistant Acinetobacter Baumannii. mSystems 2019, 4, e00068-19. [Google Scholar] [CrossRef] [Green Version]
- Finstrlová, A.; Mašlaňová, I.; Blasdel Reuter, B.G.; Doškař, J.; Götz, F.; Pantůček, R. Global Transcriptomic Analysis of Bacteriophage-Host Interactions between a Kayvirus Therapeutic Phage and Staphylococcus Aureus. Microbiol. Spectr. 2022, 10, e00123-22. [Google Scholar] [CrossRef]
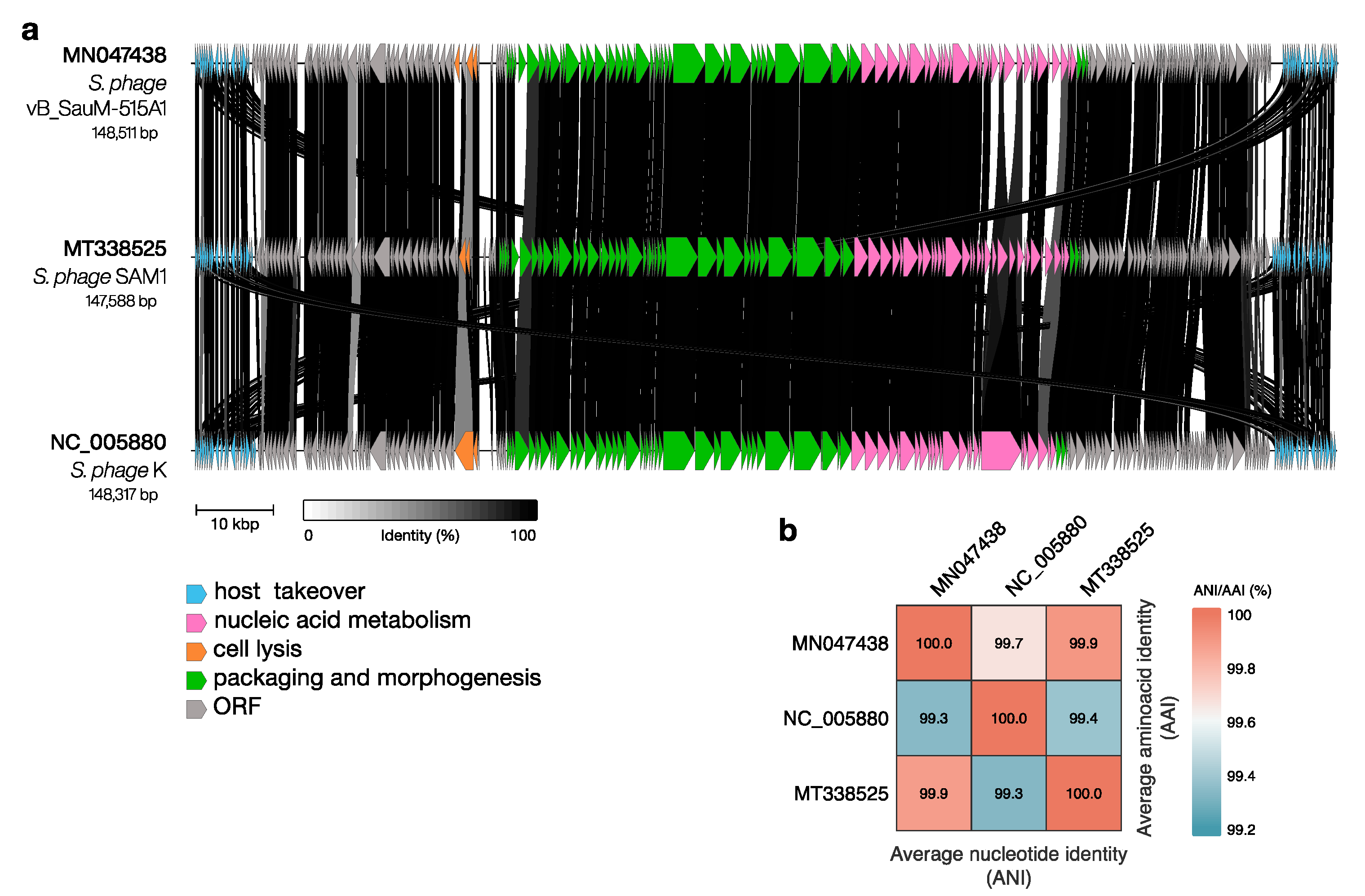
- Kornienko, M.; Fisunov, G.; Bespiatykh, D.; Kuptsov, N.; Gorodnichev, R.; Klimina, K.; Kulikov, E.; Ilina, E.; Letarov, A.; Shitikov, E. Transcriptional Landscape of Staphylococcus Aureus Kayvirus Bacteriophage VB_SauM-515A1. Viruses 2020, 12, 1320. [Google Scholar] [CrossRef]
- Arroyo-Moreno, S.; Buttimer, C.; Bottacini, F.; Chanishvili, N.; Ross, P.; Hill, C.; Coffey, A. Insights into Gene Transcriptional Regulation of Kayvirus Bacteriophages Obtained from Therapeutic Mixtures. Viruses 2022, 14, 626. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The Viral Proteomic Tree Server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [Green Version]
- Yu, G. Data Integration, Manipulation and Visualization of Phylogenetic Trees, 1st ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 2022; ISBN 978-1-00-327924-2. [Google Scholar]
- Xu, S.; Dai, Z.; Guo, P.; Fu, X.; Liu, S.; Zhou, L.; Tang, W.; Feng, T.; Chen, M.; Zhan, L.; et al. GgtreeExtra: Compact Visualization of Richly Annotated Phylogenetic Data. Mol. Biol. Evol. 2021, 38, 4039–4042. [Google Scholar] [CrossRef]
- Cui, Z.; Guo, X.; Feng, T.; Li, L. Exploring the Whole Standard Operating Procedure for Phage Therapy in Clinical Practice. J. Transl. Med. 2019, 17, 373. [Google Scholar] [CrossRef]
- Harper, D. Criteria for Selecting Suitable Infectious Diseases for Phage Therapy. Viruses 2018, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornienko, M.; Bespiatykh, D.; Malakhova, M.; Gorodnichev, R.; Kuptsov, N.; Shitikov, E. PCR Assay for Rapid Taxonomic Differentiation of Virulent Staphylococcus Aureus and Klebsiella Pneumoniae Bacteriophages. Int. J. Mol. Sci. 2023, 24, 4483. [Google Scholar] [CrossRef]
- Deghorain, M.; Van Melderen, L. The Staphylococci Phages Family: An Overview. Viruses 2012, 4, 3316–3335. [Google Scholar] [CrossRef] [Green Version]
- Azam, A.H.; Tanji, Y. Peculiarities of Staphylococcus Aureus Phages and Their Possible Application in Phage Therapy. Appl. Microbiol. Biotechnol. 2019, 103, 4279–4289. [Google Scholar] [CrossRef] [PubMed]
- Kornienko, M.; Kuptsov, N.; Gorodnichev, R.; Bespiatykh, D.; Guliaev, A.; Letarova, M.; Kulikov, E.; Veselovsky, V.; Malakhova, M.; Letarov, A.; et al. Contribution of Podoviridae and Myoviridae Bacteriophages to the Effectiveness of Anti-Staphylococcal Therapeutic Cocktails. Sci. Rep. 2020, 10, 18612. [Google Scholar] [CrossRef]
- Leskinen, K.; Tuomala, H.; Wicklund, A.; Horsma-Heikkinen, J.; Kuusela, P.; Skurnik, M.; Kiljunen, S. Characterization of VB_SauM-FRuSau02, a Twort-Like Bacteriophage Isolated from a Therapeutic Phage Cocktail. Viruses 2017, 9, 258. [Google Scholar] [CrossRef]
- McCallin, S.; Sarker, S.A.; Sultana, S.; Oechslin, F.; Brüssow, H. Metagenome Analysis of Russian and Georgian Pyophage Cocktails and a Placebo-Controlled Safety Trial of Single Phage versus Phage Cocktail in Healthy Staphylococcus aureus Carriers: Eastern Phage Cocktails against S. Aureus. Environ. Microbiol. 2018, 20, 3278–3293. [Google Scholar] [CrossRef] [PubMed]
- Taati Moghadam, M.; Khoshbayan, A.; Chegini, Z.; Farahani, I.; Shariati, A. Bacteriophages, A New Therapeutic Solution for Inhibiting Multidrug-Resistant Bacteria Causing Wound Infection: Lesson from Animal Models and Clinical Trials. Drug Des. Devel. Ther. 2020, 14, 1867–1883. [Google Scholar] [CrossRef]
- Zhou, W.-Y.; Wen, H.; Li, Y.-J.; Gao, Y.-J.; Zheng, X.-F.; Li, H.-X.; Zhu, G.-Q.; Zhang, Z.-W.; Yang, Z.-Q. WGS Analysis of Two Staphylococcus Aureus Bacteriophages from Sewage in China Provides Insights into the Genetic Feature of Highly Efficient Lytic Phages. Microbiol. Res. 2023, 271, 127369. [Google Scholar] [CrossRef] [PubMed]
- Łobocka, M.; Hejnowicz, M.S.; Dąbrowski, K.; Gozdek, A.; Kosakowski, J.; Witkowska, M.; Ulatowska, M.I.; Weber-Dąbrowska, B.; Kwiatek, M.; Parasion, S.; et al. Genomics of Staphylococcal Twort-like Phages—Potential Therapeutics of the Post-Antibiotic Era. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2012; Volume 83, pp. 143–216. ISBN 978-0-12-394438-2. [Google Scholar]
- Nováček, J.; Šiborová, M.; Benešík, M.; Pantůček, R.; Doškař, J.; Plevka, P. Structure and Genome Release of Twort-like Myoviridae Phage with a Double-Layered Baseplate. Proc. Natl. Acad. Sci. USA 2016, 113, 9351–9356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajuebor, J.; Buttimer, C.; Arroyo-Moreno, S.; Chanishvili, N.; Gabriel, E.; O’Mahony, J.; McAuliffe, O.; Neve, H.; Franz, C.; Coffey, A. Comparison of Staphylococcus Phage K with Close Phage Relatives Commonly Employed in Phage Therapeutics. Antibiotics 2018, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Rountree, P.M. The Serological Differentiation of Staphylococcal Bacteriophages. J. Gen. Microbiol. 1949, 3, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap.Js: Automatic Generation of Gene Cluster Comparison Figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Park, S.; Chun, J. Introducing EzAAI: A Pipeline for High Throughput Calculations of Prokaryotic Average Amino Acid Identity. J. Microbiol. 2021, 59, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Kuptsov, N.; Kornienko, M.; Bespiatykh, D.; Gorodnichev, R.; Klimina, K.; Veselovsky, V.; Shitikov, E. Global Transcriptomic Response of Staphylococcus Aureus to Virulent Bacteriophage Infection. Viruses 2022, 14, 567. [Google Scholar] [CrossRef]
- O’Flaherty, S.; Coffey, A.; Edwards, R.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. Genome of Staphylococcal Phage K: A New Lineage of Myoviridae Infecting Gram-Positive Bacteria with a Low G+C Content. J. Bacteriol. 2004, 186, 2862–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botka, T.; Pantůček, R.; Mašlaňová, I.; Benešík, M.; Petráš, P.; Růžičková, V.; Havlíčková, P.; Varga, M.; Žemličková, H.; Koláčková, I.; et al. Lytic and Genomic Properties of Spontaneous Host-Range Kayvirus Mutants Prove Their Suitability for Upgrading Phage Therapeutics against Staphylococci. Sci. Rep. 2019, 9, 5475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandersteegen, K.; Mattheus, W.; Ceyssens, P.-J.; Bilocq, F.; De Vos, D.; Pirnay, J.-P.; Noben, J.-P.; Merabishvili, M.; Lipinska, U.; Hermans, K.; et al. Microbiological and Molecular Assessment of Bacteriophage ISP for the Control of Staphylococcus Aureus. PLoS ONE 2011, 6, e24418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagih, O. Ggseqlogo: A Versatile R Package for Drawing Sequence Logos. Bioinformatics 2017, 33, 3645–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilke, C.O.; Wickham, H.; Wilke, M.C.O. Package ‘Cowplot’. In Streamlined Plot Theme and Plot Annotations for ‘ggplot2; 2019; Volume 1. Available online: https://CRAN.R-project.org/package=cowplot (accessed on 21 June 2023).
- Choe, D.; Szubin, R.; Dahesh, S.; Cho, S.; Nizet, V.; Palsson, B.; Cho, B.-K. Genome-Scale Analysis of Methicillin-Resistant Staphylococcus Aureus USA300 Reveals a Tradeoff between Pathogenesis and Drug Resistance. Sci. Rep. 2018, 8, 2215. [Google Scholar] [CrossRef] [Green Version]
- Jeng, S.T.; Gardner, J.F.; Gumport, R.I. Transcription Termination by Bacteriophage T7 RNA Polymerase at Rho-Independent Terminators. J. Biol. Chem. 1990, 265, 3823–3830. [Google Scholar] [CrossRef]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. In Statistical Genomics; Springer Science+Business Media: New York, NY, USA, 2016; Volume 1418, ISBN 978-1-4939-3576-5. [Google Scholar]
- Belley, A.; Callejo, M.; Arhin, F.; Dehbi, M.; Fadhil, I.; Liu, J.; McKay, G.; Srikumar, R.; Bauda, P.; Ha, N.; et al. Competition of Bacteriophage Polypeptides with Native Replicase Proteins for Binding to the DNA Sliding Clamp Reveals a Novel Mechanism for DNA Replication Arrest in Staphylococcus Aureus. Mol. Microbiol. 2006, 62, 1132–1143. [Google Scholar] [CrossRef]
- Osmundson, J.; Montero-Diez, C.; Westblade, L.F.; Hochschild, A.; Darst, S.A. Promoter-Specific Transcription Inhibition in Staphylococcus Aureus by a Phage Protein. Cell 2012, 151, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Osmundson, J.; Darst, S.A. Biochemical Insights into the Function of Phage G1 Gp67 in Staphylococcus Aureus. Bacteriophage 2013, 3, e24767. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, R.; Vandersteegen, K. Group I Introns in Staphylococcus Bacteriophages. Future Virol. 2013, 8, 997–1005. [Google Scholar] [CrossRef]
- Blasdel, B.G.; Chevallereau, A.; Monot, M.; Lavigne, R.; Debarbieux, L. Comparative Transcriptomics Analyses Reveal the Conservation of an Ancestral Infectious Strategy in Two Bacteriophage Genera. ISME J. 2017, 11, 1988–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, Z.; Lünse, C.E.; Corbino, K.A.; Ames, T.D.; Nelson, J.W.; Roth, A.; Perkins, K.R.; Sherlock, M.E.; Breaker, R.R. Detection of 224 Candidate Structured RNAs by Comparative Analysis of Specific Subsets of Intergenic Regions. Nucleic Acids Res. 2017, 45, 10811–10823. [Google Scholar] [CrossRef] [Green Version]
- Cousin, F.J.; Lynch, D.B.; Chuat, V.; Bourin, M.J.B.; Casey, P.G.; Dalmasso, M.; Harris, H.M.B.; McCann, A.; O’Toole, P.W. A Long and Abundant Non-Coding RNA in Lactobacillus salivarius. Microb. Genomics 2017, 3, e000126. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.A.; Breaker, R.R. Large Noncoding RNAs in Bacteria. Microbiol. Spectr. 2018, 6, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Lysis from Without. Bacteriophage 2011, 1, 46–49. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, C.; Shen, W.; Huang, G.; Le, S.; Lu, S.; Li, M.; Zhao, Y.; Wang, J.; Rao, X.; et al. Global Transcriptomic Analysis of Interactions between Pseudomonas Aeruginosa and Bacteriophage PaP3. Sci. Rep. 2016, 6, 19237. [Google Scholar] [CrossRef] [Green Version]
- Nanda, A.M.; Thormann, K.; Frunzke, J. Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J. Bacteriol. 2015, 197, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Dakheel, K.H.; Abdul Rahim, R.; Al-Obaidi, J.R.; Neela, V.K.; Hun, T.G.; Mat Isa, M.N.; Razali, N.; Yusoff, K. Proteomic Analysis Revealed the Biofilm-Degradation Abilities of the Bacteriophage UPMK_1 and UPMK_2 against Methicillin-Resistant Staphylococcus Aureus. Biotechnol. Lett. 2022, 44, 513–522. [Google Scholar] [CrossRef]
- Bleriot, I.; Blasco, L.; Pacios, O.; Fernández-García, L.; López, M.; Ortiz-Cartagena, C.; Barrio-Pujante, A.; Fernández-Cuenca, F.; Pascual, Á.; Martínez-Martínez, L.; et al. Proteomic Study of the Interactions between Phages and the Bacterial Host Klebsiella pneumoniae. Microbiol. Spectr. 2023, 11, e03974-22. [Google Scholar] [CrossRef]
- Papaianni, M.; Cuomo, P.; Fulgione, A.; Albanese, D.; Gallo, M.; Paris, D.; Motta, A.; Iannelli, D.; Capparelli, R. Bacteriophages Promote Metabolic Changes in Bacteria Biofilm. Microorganisms 2020, 8, 480. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Duration of Adsorption Period (Adsorption of More than 60% of Phage Particles), Min | Duration of the Latent Period, Min | Time Points for Transcriptional Analysis, Min | References |
|---|---|---|---|---|
| vB_SauM-515A1 | 5 | 30–40 | 5, 15, 30 | [30] |
| K | 2 | 30–35 | 2, 5, 10, 20, 30 | [29] |
| SAM1 | NA 1 | 50 | 15, 35, 45 | [31] |
| Strain | Strain Type | MLST Type | Genome Size, Mb (GenBank Number) | CDS | Number of DEG | References |
|---|---|---|---|---|---|---|
| SA515 | clinical isolate | ST8 | 2.66 (JAKRSL000000000) | 2658 | 263 (FC ≥ |2|; FDR < 0.001) | [53] |
| E1185(IV)ST12 | clinical isolate | most similar to ST12 | 2.78 (CP089586) | 2704 | 829 (p < 0.05) | [29] |
| Newman | laboratory strain | ST8 | 2.89 (NC_009641.1) | 2614 | 625 (FC ≥ |1.5|; FDR < 0.001) | [31] |
| SH1000 | laboratory strain is a derivative of strain NCTC8325, characterized by the absence of prophage and RsbU repair | ST8 | 2.68 (JAJAFP000000000) | 2684 | 150 (FC ≥ |1.5|; FDR < 0.001) |
| Virulence Factor | Gene | References |
|---|---|---|
| Toxin | ||
| alpha hemolysin | hlgy | [29,31] |
| gamma hemolysin subunit A | hlgA | [29] |
| gamma hemolysin subunit CB | hlgB, hlgC | [29,31,53] |
| superantigen-like protein, exotoxin | set15 | [53] |
| Adhesins | ||
| fibrinogen-binding protein | efb | [53] |
| fibronectin-binding protein FnbA | fnbA | [31] |
| fibronectin-binding protein FnbB | fnbB | [29] |
| MSCRAMM family adhesin clumping factor ClfA | clfA | [29] |
| extracellular adherence protein Eap/Map | eap | [31] |
| Immune system evasion | ||
| immunoglobulin-binding protein | sbi | [29,31,53] |
| staphylococcal complement inhibitor SCIN | scn | [31,53] |
| chemotaxis inhibiting protein Chp | chp | [29] |
| Virulence factors associated with secretion | ||
| ESAT-6/WXG100 family secreted protein EsxA/YukE | esxA | [53] |
| protein secretion system EssA | essA | [53] |
| Strain | Most Similar Phage | Genus | Integrase Type | Hyperexpression |
|---|---|---|---|---|
| SA515 | phi2958PVL (NC_011344) | Triavirus | Sa2 | − |
| phiNM4 (NC_028864.1) | Phietavirus | Sa7 | − | |
| P282 (NC_048634.1) | Biseptimavirus | Sa3 | + | |
| E1185(IV)ST12 | 3MRA (NC_028917) | Phietavirus | Sa5 | + |
| Newman | phiNM1 (NC_008583.1) | Dubowvirus | Sa5 | + |
| phiNM2 (NC_028913.1) | Dubowvirus | Sa7 | + | |
| phiNM3 (NC_008617.1) | Biseptimavirus | Sa3 | + | |
| phiNM4 (NC_028864.1) | Phietavirus | Sa6 | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kornienko, M.; Bespiatykh, D.; Gorodnichev, R.; Abdraimova, N.; Shitikov, E. Transcriptional Landscapes of Herelleviridae Bacteriophages and Staphylococcus aureus during Phage Infection: An Overview. Viruses 2023, 15, 1427. https://doi.org/10.3390/v15071427
Kornienko M, Bespiatykh D, Gorodnichev R, Abdraimova N, Shitikov E. Transcriptional Landscapes of Herelleviridae Bacteriophages and Staphylococcus aureus during Phage Infection: An Overview. Viruses. 2023; 15(7):1427. https://doi.org/10.3390/v15071427
Chicago/Turabian StyleKornienko, Maria, Dmitry Bespiatykh, Roman Gorodnichev, Narina Abdraimova, and Egor Shitikov. 2023. "Transcriptional Landscapes of Herelleviridae Bacteriophages and Staphylococcus aureus during Phage Infection: An Overview" Viruses 15, no. 7: 1427. https://doi.org/10.3390/v15071427
APA StyleKornienko, M., Bespiatykh, D., Gorodnichev, R., Abdraimova, N., & Shitikov, E. (2023). Transcriptional Landscapes of Herelleviridae Bacteriophages and Staphylococcus aureus during Phage Infection: An Overview. Viruses, 15(7), 1427. https://doi.org/10.3390/v15071427