
Mining Public Data to Investigate the Virome of Neglected Pollinators and Other Floral Visitors
 , , ,
, , ,  , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Species Selection
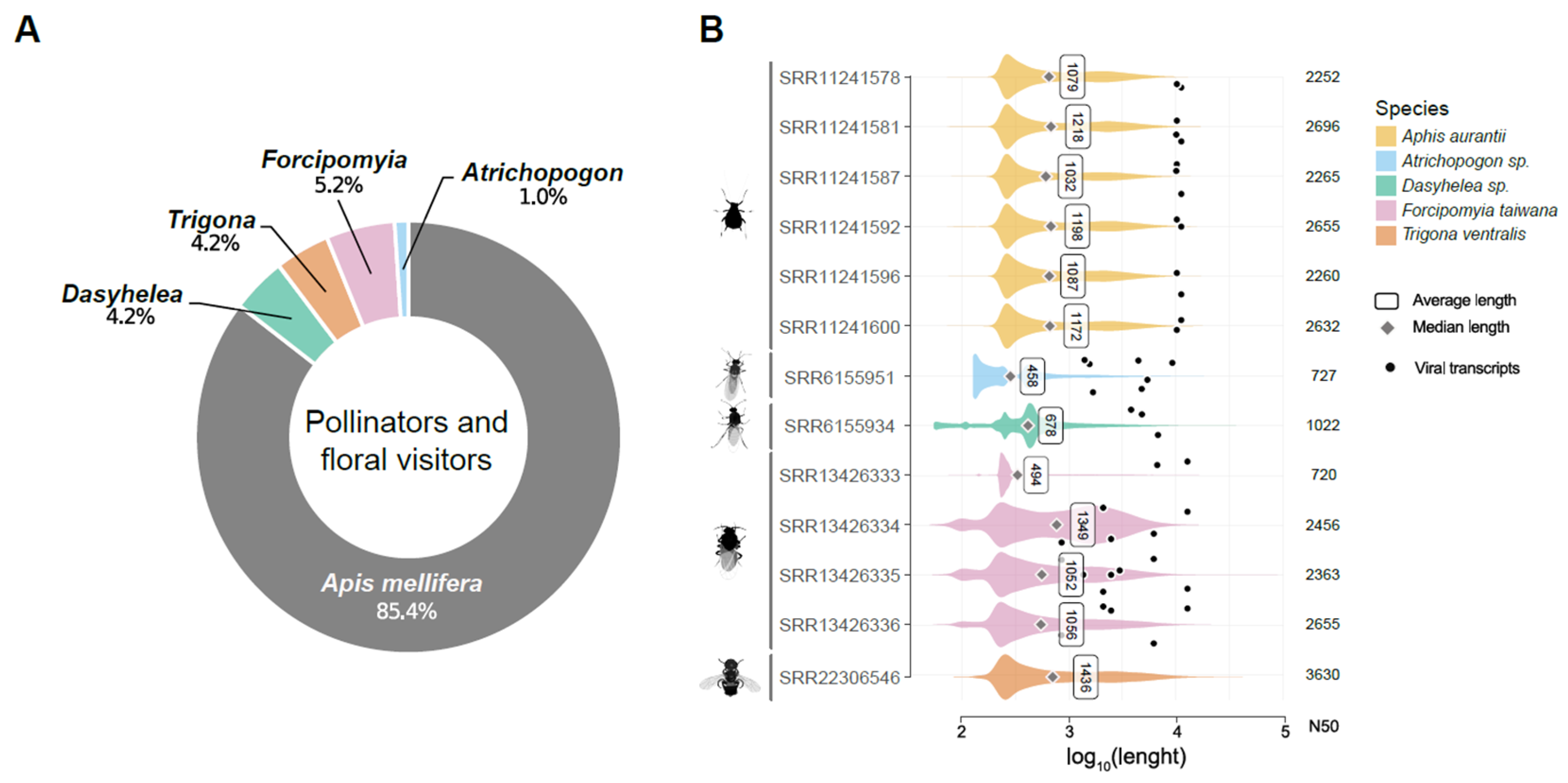
2.2. Retrieval of RNA-Seq Libraries
2.3. Bioinformatic Analysis
2.3.1. Pre-Processing, Integrative Assembly, and Viral Sequences Identification
2.3.2. Manual Curation
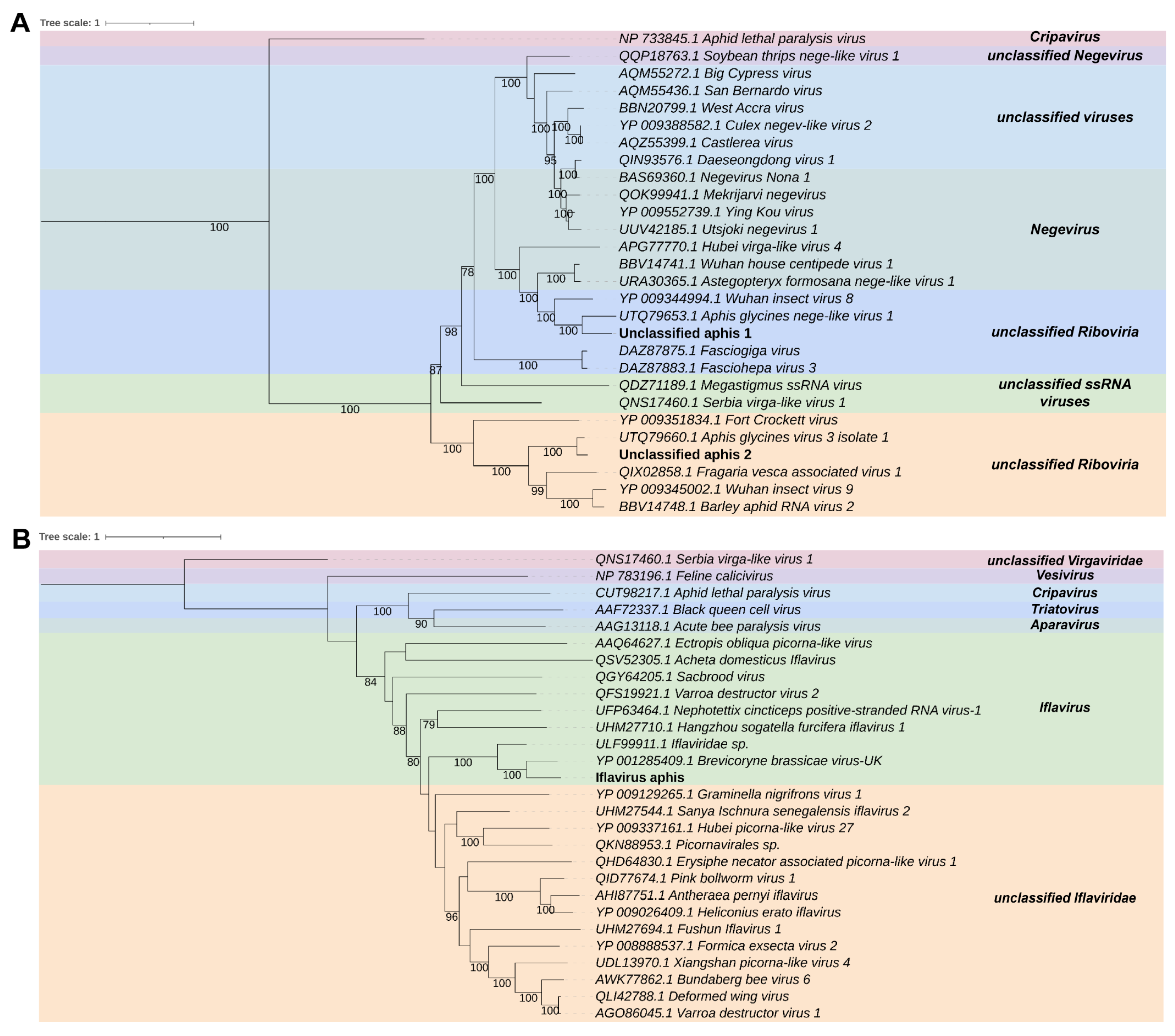
2.4. Phylogenetic Analysis
2.5. Abundance of Viral Sequences
3. Results
3.1. Metatranscriptome Assembly
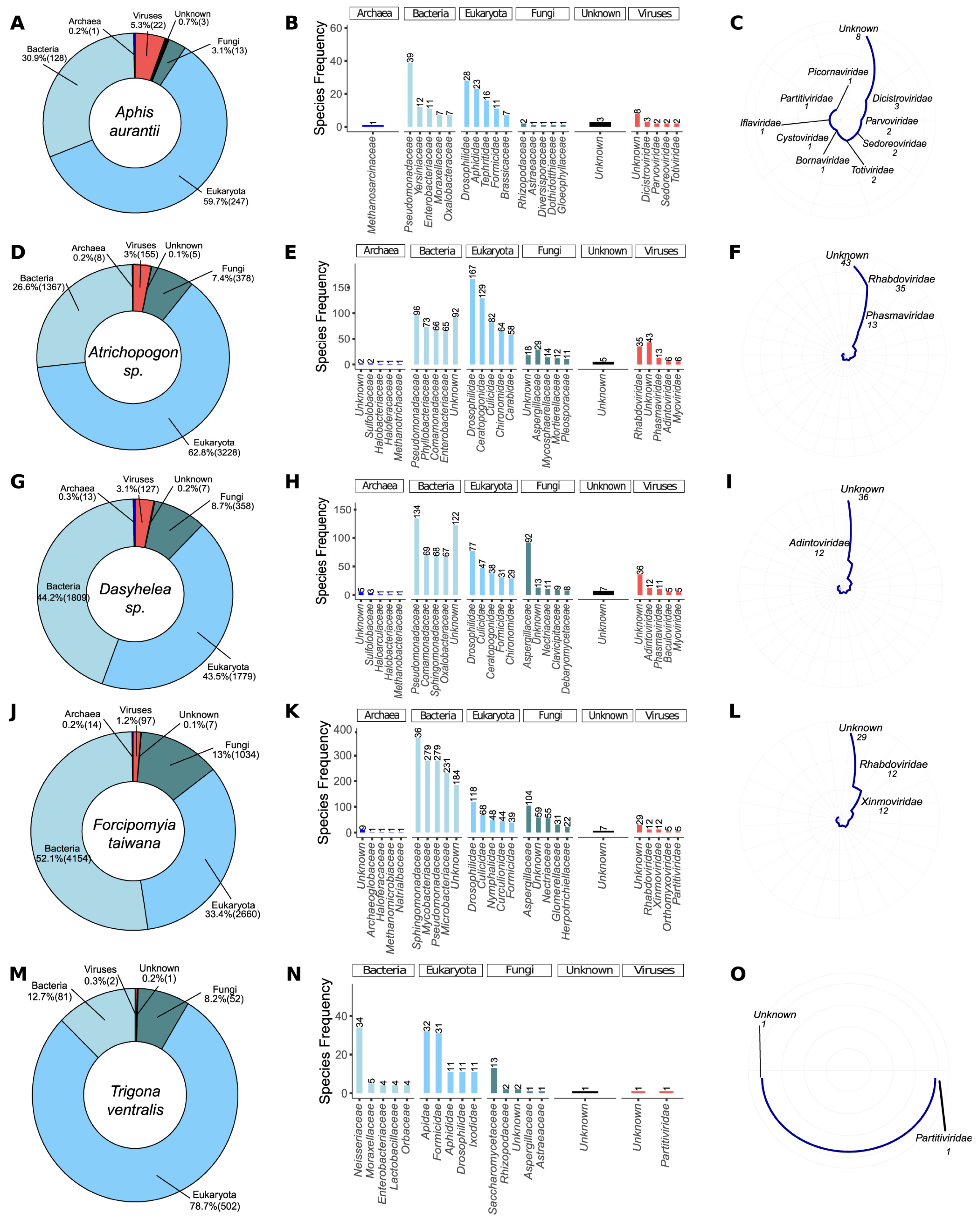
3.2. Metagenomic Analysis
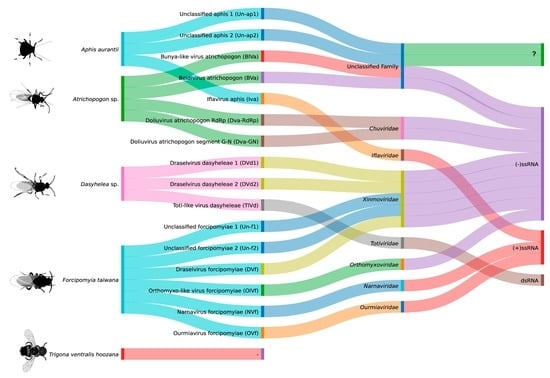
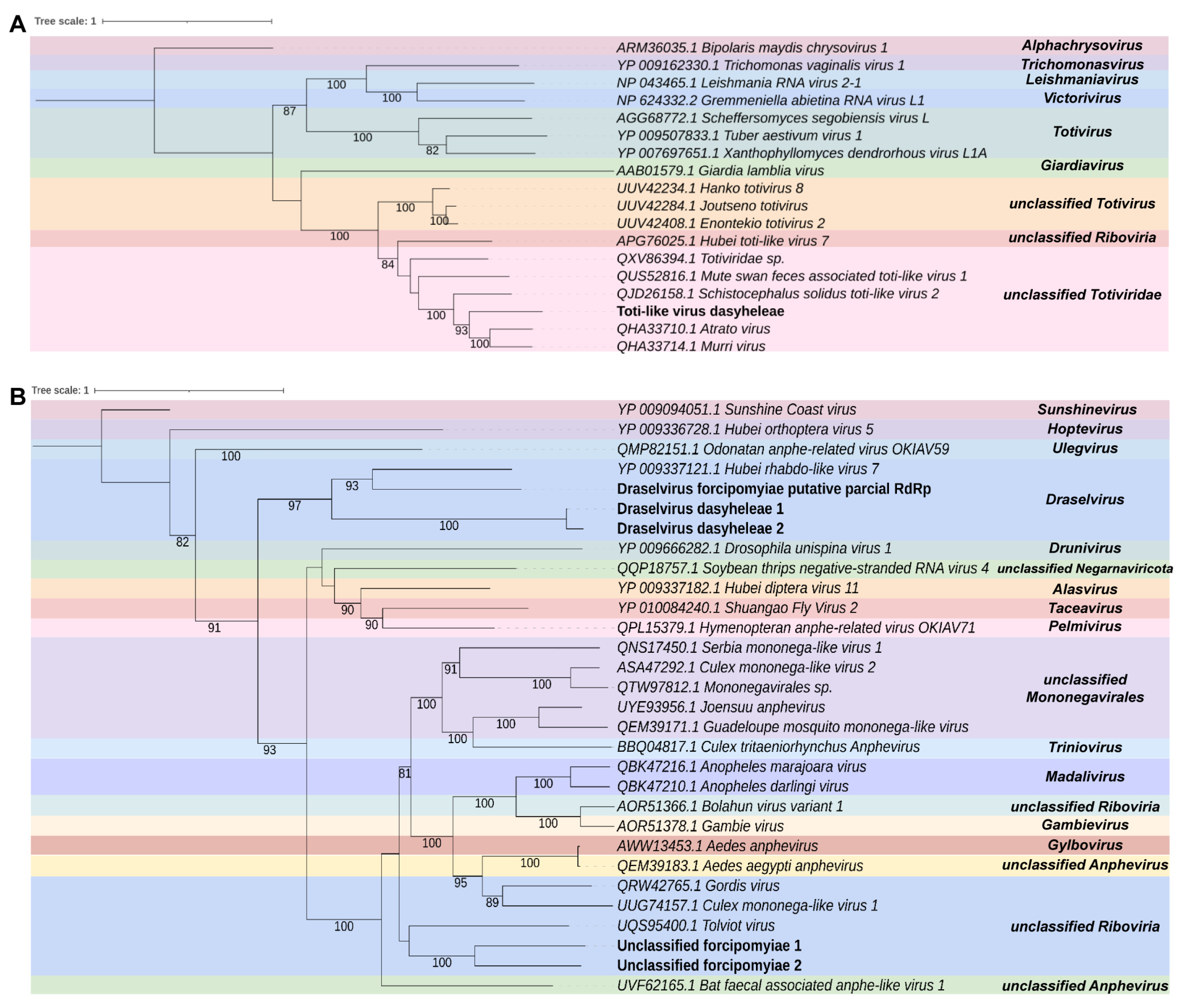
3.3. Virome Characterization
- Aphis aurantii
- Atrichopogon sp.
- Dasyhelea sp.
- Forcipomyia taiwana
3.4. Abundance of Viral Sequences
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ollerton, J.; Winfree, R.; Tarrant, S. How many flowering plants are pollinated by animals? Oikos 2011, 120, 321–326. [Google Scholar] [CrossRef]
- Kearns, C.A.; Inouye, D.W.; Waser, N.M. ENDANGERED MUTUALISMS: The Conservation of Plant-Pollinator Interactions. Annu. Rev. Ecol. Syst. 1998, 29, 83–112. [Google Scholar] [CrossRef]
- Klein, A.-M.; Vaissière, B.E.; Cane, J.H.; Steffan-Dewenter, I.; Cunningham, S.A.; Kremen, C.; Tscharntke, T. Importance of pollinators in changing landscapes for world crops. Proc. R. Soc. B Biol. Sci. 2007, 274, 303–313. [Google Scholar] [CrossRef]
- Potts, S.G.; Biesmeijer, J.C.; Kremen, C.; Neumann, P.; Schweiger, O.; Kunin, W.E. Global pollinator declines: Trends, impacts and drivers. Trends Ecol. Evol. 2010, 25, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Winfree, R.; Aguilar, R.; Vázquez, D.P.; Lebuhn, G.; Aizen, M.A. A meta-analysis of bees’ responses to anthropogenic disturbance. Ecology 2009, 90, 2068–2076. [Google Scholar] [CrossRef]
- Saunders, M.E. Insect pollinators collect pollen from wind-pollinated plants: Implications for pollination ecology and sustainable agriculture. Insect Conserv. Divers. 2018, 11, 13–31. [Google Scholar] [CrossRef]
- Tian, L.; Ren, J.; Li, R.; Di, N.; Huang, X.; Wang, S.; Fang, X.; Xu, X. The Honeybee (Apis mellifera L.) Is an Efficient Pollinator for Paeonia lactiflora Pall in the Field. Appl. Sci. 2023, 13, 1179. [Google Scholar] [CrossRef]
- Rizzardo, R.A.; Milfont, M.O.; da Silva, E.M.; Freitas, B.M. Apis mellifera pollination improves agronomic productivity of anemophilous castor bean (Ricinus communis). An. Acad. Bras. Ciênc. 2012, 84, 1137–1145. [Google Scholar] [CrossRef]
- Suganthy, M.; Krishna, K.G.; Kumar, S.M.; Jegadeeswari, V. Indian pollinators of cocoa, Theobroma cacao L. Acta Hortic. 2019, 451–458. [Google Scholar] [CrossRef]
- Montero-Cedeño, S.; Cañarte-Bermudez, E.; Navarrete-Cedeño, J.; Pinargote-Borrero, A.; Sanchez-Hernández, P. Ceratopogonidae: Their role in pollination and fertilization at various technological levels of Theobroma cacao L. production. Rev. Fac. Agron. Univ. Zulia. 2022, 39, e223943. [Google Scholar] [CrossRef]
- Peach, D.A.H.; Gries, G. Mosquito phytophagy—Sources exploited, ecological function, and evolutionary transition to haematophagy. Entomol. Exp. Appl. 2020, 168, 120–136. [Google Scholar] [CrossRef]
- Requier, F.; Pérez-Méndez, N.; Andersson, G.K.; Blareau, E.; Merle, I.; Garibaldi, L.A. Bee and non-bee pollinator importance for local food security. Trends Ecol. Evol. 2023, 38, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Brosi, B.J.; Daily, G.C.; Ehrlich, P.R. Bee Community Shifts with Landscape Context in a Tropical Countryside. Ecol. Appl. 2007, 17, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Rader, R.; Bartomeus, I.; Garibaldi, L.A.; Garratt, M.P.D.; Howlett, B.G.; Winfree, R.; Cunningham, S.A.; Mayfield, M.M.; Arthur, A.D.; Andersson, G.K.S.; et al. Non-bee insects are important contributors to global crop pollination. Proc. Natl. Acad. Sci. USA 2016, 113, 146–151. [Google Scholar] [CrossRef] [PubMed]
- McArt, S.H.; Koch, H.; Irwin, R.E.; Adler, L.S. Arranging the bouquet of disease: Floral traits and the transmission of plant and animal pathogens. Ecol. Lett. 2014, 17, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Shilts, T.; El-Mohtar, C.; Dawson, W.O.; Killiny, N. Citrus tristeza virus P33 Protein Is Required for Efficient Transmission by the Aphid Aphis (Toxoptera) citricidus (Kirkaldy). Viruses 2020, 12, 1131. [Google Scholar] [CrossRef]
- Rader, R.; Cunningham, S.; Howlett, B.; Inouye, D. Non-Bee Insects as Visitors and Pollinators of Crops: Biology, Ecology, and Management. Annu. Rev. Entomol. 2020, 65, 391–407. [Google Scholar] [CrossRef]
- Gallai, N.; Salles, J.-M.; Settele, J.; Vaissière, B.E. Economic valuation of the vulnerability of world agriculture confronted with pollinator decline. Ecol. Econ. 2009, 68, 810–821. [Google Scholar] [CrossRef]
- Biesmeijer, J.C.; Roberts, S.P.M.; Reemer, M.; Ohlemüller, R.; Edwards, M.; Peeters, T.; Schaffers, A.P.; Potts, S.G.; Kleukers, R.J.M.C.; Thomas, C.D.; et al. Parallel Declines in Pollinators and Insect-Pollinated Plants in Britain and the Netherlands. Science 2006, 313, 351–354. [Google Scholar] [CrossRef]
- Burkle, L.A.; Marlin, J.C.; Knight, T.M. Plant-Pollinator Interactions over 120 Years: Loss of Species, Co-Occurrence, and Function. Science 2013, 339, 1611–1615. [Google Scholar] [CrossRef]
- Winder, J.A. Field observations on Ceratopogonidae and other Diptera: Nematocera associated with cocoa flowers in Brazil. Bull. Entomol. Res. 1977, 67, 57–63. [Google Scholar] [CrossRef]
- Warmke, H.E. Studies on Natural Pollination of Hevea brasiliensis in Brazil. Science 1952, 116, 474–475. [Google Scholar] [CrossRef]
- Narayanan Kutty, S.; Wong, W.H.; Meusemann, K.; Meier, R.; Cranston, P.S. A phylogenomic analysis of Culicomorpha (Diptera) resolves the relationships among the eight constituent families: Phylogenomic analysis of Culicomorpha. Syst. Entomol. 2018, 43, 434–446. [Google Scholar] [CrossRef]
- Steiner, K.E. Functional Dioecism in the Malpighiaceae: The Breeding System of Spachea membranacea Cuatr. Am. J. Bot. 1985, 72, 1537–1543. [Google Scholar] [CrossRef]
- Panahi, E.; Shivas, M.; Hall-Mendelin, S.; Kurucz, N.; Rudd, P.A.; De Araujo, R.; Skinner, E.B.; Melville, L.; Herrero, L.J. Utilising a novel surveillance system to investigate species of Forcipomyia (Lasiohelea) (Diptera: Ceratopogonidae) as the suspected vectors of Leishmania macropodum (Kinetoplastida: Trypanosomatidae) in the Darwin region of Australia. Int. J. Parasitol. Parasites Wildl. 2020, 12, 192–198. [Google Scholar] [CrossRef]
- Pinheiro, F.P.; Travassos, A.P.A.; Gomes, M.L.C.; LeDuc, J.W.; Hoch, A.L. Transmission of Oropouche Virus from Man to Hamster by the Midge Culicoides paraensis. Science 1982, 215, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- McFrederick, Q.S.; Wcislo, W.T.; Hout, M.C.; Mueller, U.G. Host species and developmental stage, but not host social structure, affects bacterial community structure in socially polymorphic bees. FEMS Microbiol. Ecol. 2014, 88, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Beard, C.B.; Mason, P.W.; Aksoy, S.; Tesh, R.B.; Richards, F.F. Transformation of an Insect Symbiont and Expression of a Foreign Gene in the Chagas’ Disease Vector Rhodnius Prolixus. Am. J. Trop. Med. Hyg. 1992, 46, 195–200. [Google Scholar] [CrossRef]
- Jia, D.; Mao, Q.; Chen, Y.; Liu, Y.; Chen, Q.; Wu, W.; Zhang, X.; Chen, H.; Li, Y.; Wei, T. Insect symbiotic bacteria harbour viral pathogens for transovarial transmission. Nat. Microbiol. 2017, 2, 17025. [Google Scholar] [CrossRef]
- Roossinck, B.J.; Papanicolaou, M.; García-Arenal, F. Ecosystem simplification, biodiversity loss and plant virus emergence. Curr. Opin. Virol. 2015, 10, 56–62. [Google Scholar] [CrossRef]
- Hong, F.; Mo, S.-H.; Liu, Y.; Wei, D. Transcriptomic Profiling of Various Developmental Stages of Aphis Aurantii to Provide a Genetic Resource for Gene Expression and SSR Analysis. Front. Physiol. 2020, 11, 578939. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-E.; Tsai, M.-H.; Huang, H.-T.; Tsai, C.-C.; Chen, M.-J.; Yang, D.-S.; Yang, T.Z.; Wang, J.; Huang, R.N. Transcriptome profiling reveals the developmental regulation of NaCl-treated Forcipomyia taiwana eggs. BMC Genom. 2021, 22, 792. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High. Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 July 2023).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 2019, 8, giz100. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. SPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust. de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Ren, H. TaxonKit: A practical and efficient NCBI taxonomy toolkit. J. Genet. Genom. 2021, 48, 844–850. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA Sequence Assembly Program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.; Wurtele, E.S. orfipy: A fast and flexible tool for extracting ORFs. Bioinformatics 2021, 37, 3019–3020. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Rombel, I.T.; Sykes, K.F.; Rayner, S.; Johnston, S.A. ORF-FINDER: A vector for high-throughput gene identification. Gene 2002, 282, 33–41. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, T.; Sauvion, N.; Almeida, R.P.; Fournet, F.; Alout, H. The ecological significance of arthropod vectors of plant, animal, and human pathogens. Trends Parasitol. 2022, 38, 404–418. [Google Scholar] [CrossRef]
- Fattorini, R.; Glover, B.J. Molecular Mechanisms of Pollination Biology. Annu. Rev. Plant Biol. 2020, 71, 487–515. [Google Scholar] [CrossRef] [PubMed]
- Waser, N.M.; Chittka, L.; Price, M.V.; Williams, N.M.; Ollerton, J. Generalization in Pollination Systems, and Why it Matters. Ecology 1996, 77, 1043–1060. [Google Scholar] [CrossRef]
- Guo, Y.; Ji, N.; Bai, L.; Ma, J.; Li, Z. Aphid Viruses: A Brief View of a Long History. Front. Insect Sci. 2022, 2, 846716. [Google Scholar] [CrossRef]
- Wan, G.; Jiang, S.; Wang, W.; Li, G.; Tao, X.; Pan, W.; Sword, G.A.; Chen, F. Rice stripe virus counters reduced fecundity in its insect vector by modifying insect physiology, primary endosymbionts and feeding behavior. Sci. Rep. 2015, 5, 12527. [Google Scholar] [CrossRef]
- Sun, M.; Ji, Y.; Li, G.; Shao, J.; Chen, R.; Gong, H.; Chen, S.; Chen, J. Highly adaptive Phenuiviridae with biomedical importance in multiple fields. J. Med. Virol. 2022, 94, 2388–2401. [Google Scholar] [CrossRef]
- Liu, W.; Hajano, J.-U.; Wang, X. New insights on the transmission mechanism of tenuiviruses by their vector insects. Curr. Opin. Virol. 2018, 33, 13–17. [Google Scholar] [CrossRef]
- Kormelink, R.; Verchot, J.; Tao, X.; Desbiez, C. The Bunyavirales: The Plant-Infecting Counterparts. Viruses 2021, 13, 842. [Google Scholar] [CrossRef]
- Bratuleanu, B.E.; Temmam, S.; Munier, S.; Chrétien, D.; Bigot, T.; van der Werf, S.; Savuta, G.; Eloit, M. Detection of Phenuiviridae, Chuviridae Members, and a Novel Quaranjavirus in Hard Ticks From Danube Delta. Front. Vet. Sci. 2022, 9, 863814. [Google Scholar] [CrossRef] [PubMed]
- Asha, K.; Kumar, B. Emerging Influenza D Virus Threat: What We Know so Far! J. Clin. Med. 2019, 8, 192. [Google Scholar] [CrossRef]
- Esteban, R.; Fujimura, T. Launching the yeast 23S RNA Narnavirus shows 5′ and 3′ cis-acting signals for replication. Proc. Natl. Acad. Sci. USA 2003, 100, 2568–2573. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhou, J.; Zhou, X.; Shuai, S.; Zhou, R.; An, H.; Fang, S.; Zhang, S.; Deng, Q. A novel narnavirus from the plant-pathogenic fungus Magnaporthe oryzae. Arch. Virol. 2020, 165, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, P.; Ferreira, F.; da Silva, F.; Oliveira, L.S.; Marques, J.T.; Goes-Neto, A.; Aguiar, E.; Gruber, A. Characterization of a Novel Mitovirus of the Sand Fly Lutzomyia longipalpis Using Genomic and Virus–Host Interaction Signatures. Viruses 2020, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Turina, M.; Hillman, B.I.; Izadpanah, K.; Rastgou, M.; Rosa, C. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Ourmiavirus. J. Gen. Virol. 2017, 98, 129–130. [Google Scholar] [CrossRef]
- Rastgou, M.; Habibi, M.K.; Izadpanah, K.; Masenga, V.; Milne, R.G.; Wolf, Y.I.; Koonin, E.V.; Turina, M. Molecular characterization of the plant virus genus Ourmiavirus and evidence of inter-kingdom reassortment of viral genome segments as its possible route of origin. J. Gen. Virol. 2009, 90, 2525–2535. [Google Scholar] [CrossRef]
- Chiapello, M.; Bosco, L.; Ciuffo, M.; Ottati, S.; Salem, N.; Rosa, C.; Tavella, L.; Turina, M. Complexity and Local Specificity of the Virome Associated with Tospovirus-Transmitting Thrips Species. J. Virol. 2021, 95, e00597-21. [Google Scholar] [CrossRef]
- de Lima, J.G.S.; Teixeira, D.G.; Freitas, T.T.; Lima, J.P.M.S.; Lanza, D.C.F. Evolutionary origin of 2A-like sequences in Totiviridae genomes. Virus Res. 2019, 259, 1–9. [Google Scholar] [CrossRef]
- Maes, P.; Amarasinghe, G.K.; Ayllón, M.A.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Second update 2018. Arch Virol. 2019, 164, 1233–1244. [Google Scholar] [CrossRef]
- Manni, M.; Zdobnov, E.M. A Novel Anphevirus in Aedes albopictus Mosquitoes Is Distributed Worldwide and Interacts with the Host RNA Interference Pathway. Viruses 2020, 12, 1264. [Google Scholar] [CrossRef] [PubMed]
- Cazorla, C.G.; Campos, R.E. Ceratopogonidae (Diptera) Communities in a Protected Area Threatened by Urbanization. Neotrop. Entomol. 2020, 49, 361–368. [Google Scholar] [CrossRef]
- Züst, T.; Agrawal, A.A. Mechanisms and evolution of plant resistance to aphids. Nat. Plants 2016, 2, 15206. [Google Scholar] [CrossRef] [PubMed]
- Bonning, B.C. The Insect Virome: Opportunities and Challenges. Curr. Issues Mol. Biol. 2020, 34, 1–12. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Santana, S.F.; Santos, V.C.; Lopes, Í.S.; Porto, J.A.M.; Mora-Ocampo, I.Y.; Sodré, G.A.; Pirovani, C.P.; Góes-Neto, A.; Pacheco, L.G.C.; Fonseca, P.L.C.; et al. Mining Public Data to Investigate the Virome of Neglected Pollinators and Other Floral Visitors. Viruses 2023, 15, 1850. https://doi.org/10.3390/v15091850
de Santana SF, Santos VC, Lopes ÍS, Porto JAM, Mora-Ocampo IY, Sodré GA, Pirovani CP, Góes-Neto A, Pacheco LGC, Fonseca PLC, et al. Mining Public Data to Investigate the Virome of Neglected Pollinators and Other Floral Visitors. Viruses. 2023; 15(9):1850. https://doi.org/10.3390/v15091850
Chicago/Turabian Stylede Santana, Sabrina Ferreira, Vinícius Castro Santos, Ícaro Santos Lopes, Joel Augusto Moura Porto, Irma Yuliana Mora-Ocampo, George Andrade Sodré, Carlos Priminho Pirovani, Aristóteles Góes-Neto, Luis Gustavo Carvalho Pacheco, Paula Luize Camargos Fonseca, and et al. 2023. "Mining Public Data to Investigate the Virome of Neglected Pollinators and Other Floral Visitors" Viruses 15, no. 9: 1850. https://doi.org/10.3390/v15091850