
Development of a Replication-Deficient Bacteriophage Reporter Lacking an Essential Baseplate Wedge Subunit

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Bacteriophage Stocks
2.2. Genetic Comparison of SEA1 and T4
2.3. Design of Homologous Recombination and Complementation Plasmid
2.4. Genetic Engineering of SEA1∆gp141.NL
2.5. Phage DNA Preparation and PCR Confirmation
2.6. Evaluation of Spot Lysis and Spot Plaque Formation
2.7. One-Step Growth Curve
2.8. NanoLuc® Reporter Phage Assay
3. Results
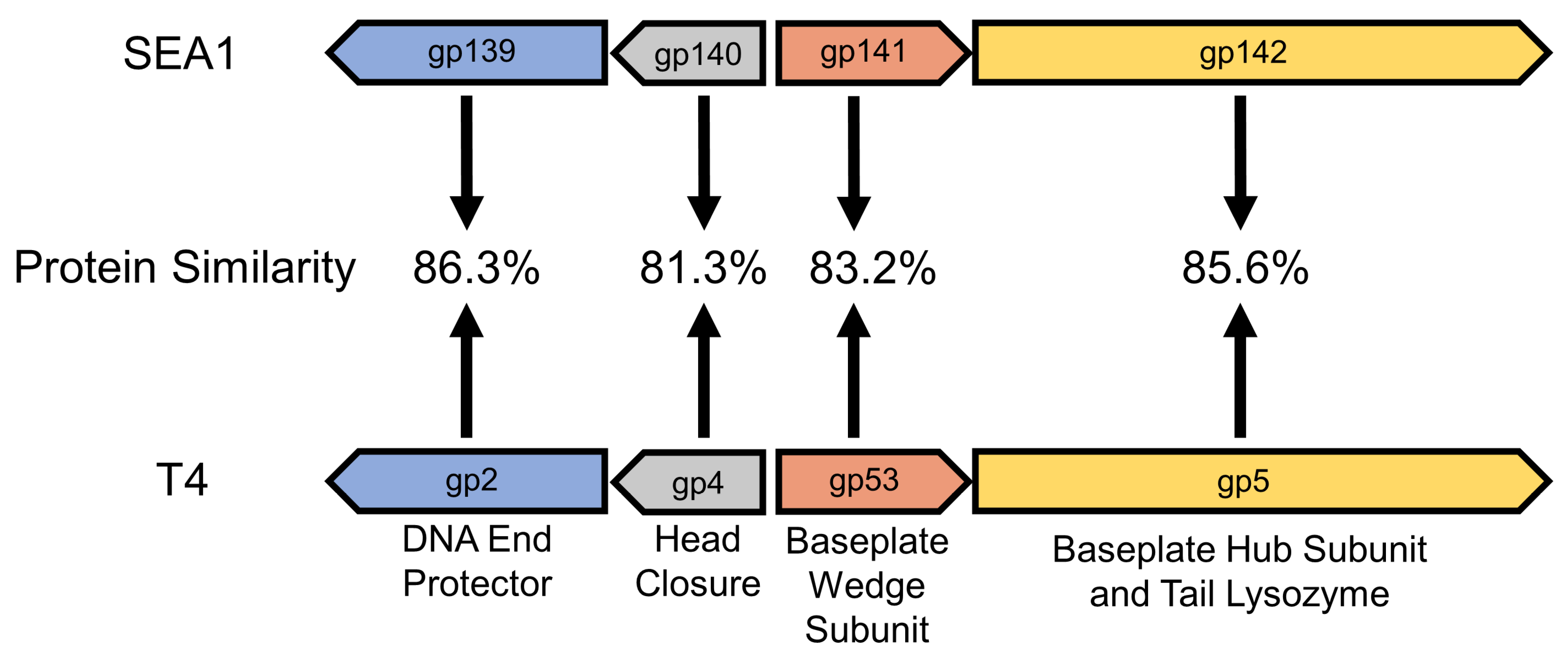
3.1. SEA1 Encodes a Homolog to gp53, T4’s Essential Baseplate Wedge Subunit
3.2. Genetic Engineering of SEA1∆gp141.NL
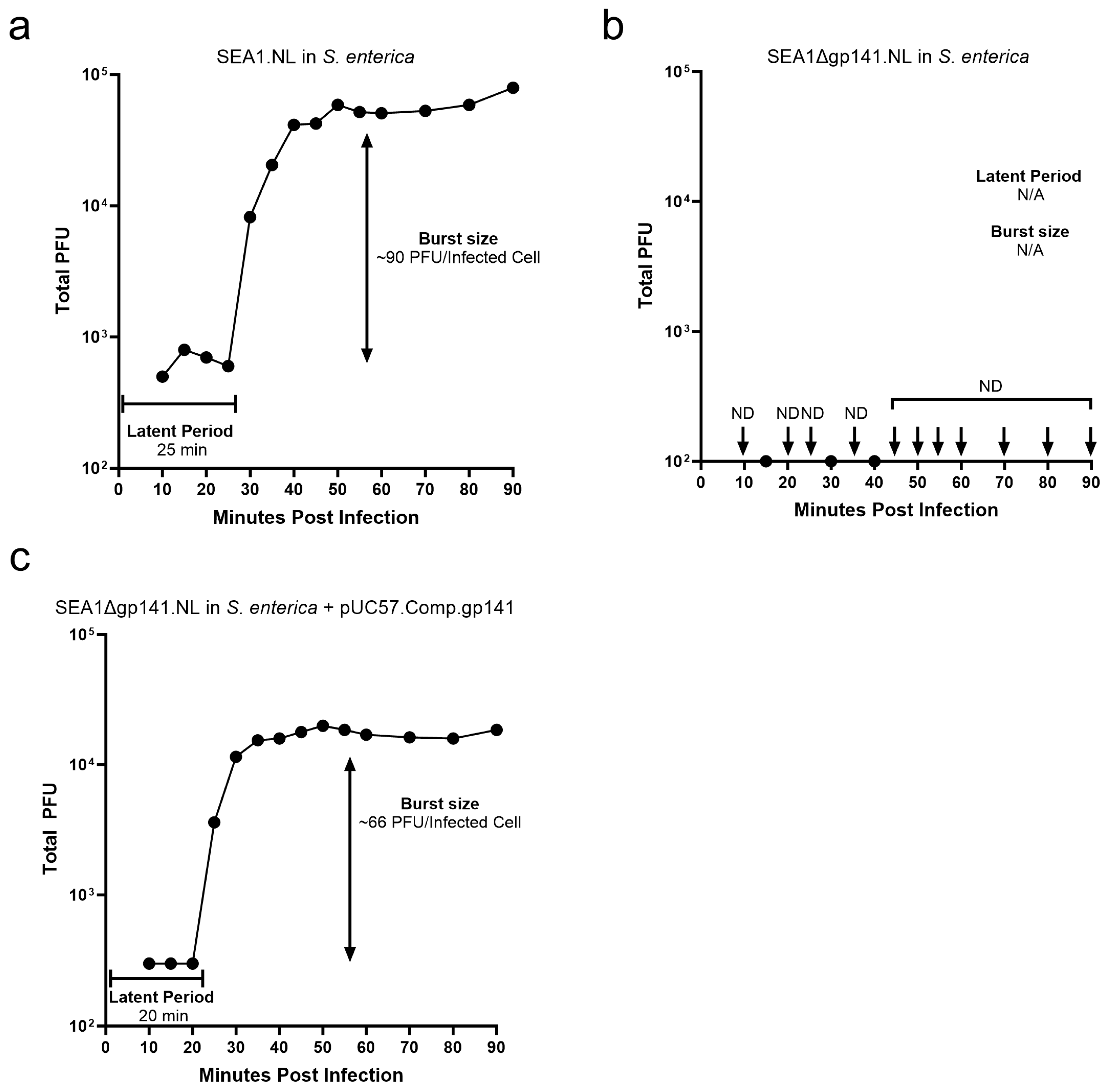
3.3. SEA1∆gp141.NL Is Deficient in Plaque Formation and Replication
3.4. Despite Replication-Deficiency, SEA1∆gp141.NL Remains an Effective Salmonella Reporter
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [PubMed]
- Selle, K.; Fletcher, J.R.; Tuson, H.; Schmitt, D.S.; McMillan, L.; Vridhambal, G.S.; Rivera, A.J.; Montgomery, S.A.; Fortier, L.C.; Barrangou, R.; et al. In Vivo Targeting of Clostridioides difficile Using Phage-Delivered CRISPR-Cas3 Antimicrobials. mBio 2020, 11, e00019-20. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Liu, Y.; Chen, Y.; Hu, J.; Xiao, W.; Tang, X.; Li, G.; Lin, P.; Pu, Q.; Wu, Q.; et al. Engineered Bacteriophages Containing Anti-CRISPR Suppress Infection of Antibiotic-Resistant P. aeruginosa. Microbiol. Spectr. 2022, 10, e0160222. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.; Prokhorov, N.S.; Loessner, M.J.; Leiman, P.G. Reprogramming bacteriophage host range: Design principles and strategies for engineering receptor binding proteins. Curr. Opin. Biotechnol. 2021, 68, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Paulson, J.; Brown, M.; Zahn, H.; Nguyen, M.M.; Eisenberg, M.; Erickson, S. Tailoring the Host Range of Ackermannviridae Bacteriophages through Chimeric Tailspike Proteins. Viruses 2023, 15, 286. [Google Scholar] [CrossRef]
- Klumpp, J.; Loessner, M.J. Detection of bacteria with bioluminescent reporter bacteriophage. Adv. Biochem. Eng. Biotechnol. 2014, 144, 155–171. [Google Scholar] [CrossRef]
- Funatsu, T.; Taniyama, T.; Tajima, T.; Tadakuma, H.; Namiki, H. Rapid and sensitive detection method of a bacterium by using a GFP reporter phage. Microbiol. Immunol. 2002, 46, 365–369. [Google Scholar] [CrossRef]
- Alcaine, S.D.; Pacitto, D.; Sela, D.A.; Nugen, S.R. Phage & phosphatase: A novel phage-based probe for rapid, multi-platform detection of bacteria. Analyst 2015, 140, 7629–7636. [Google Scholar] [CrossRef]
- Burnham, S.; Hu, J.; Anany, H.; Brovko, L.; Deiss, F.; Derda, R.; Griffiths, M.W. Towards rapid on-site phage-mediated detection of generic Escherichia coli in water using luminescent and visual readout. Anal. Bioanal. Chem. 2014, 406, 5685–5693. [Google Scholar] [CrossRef]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.M.; Gil, J.; Brown, M.; Cesar Tondo, E.; Soraya Martins de Aquino, N.; Eisenberg, M.; Erickson, S. Accurate and sensitive detection of Salmonella in foods by engineered bacteriophages. Sci. Rep. 2020, 10, 17463. [Google Scholar] [CrossRef] [PubMed]
- Meile, S.; Sarbach, A.; Du, J.; Schuppler, M.; Saez, C.; Loessner, M.J.; Kilcher, S. Engineered Reporter Phages for Rapid Bioluminescence-Based Detection and Differentiation of Viable Listeria Cells. Appl. Environ. Microbiol. 2020, 86, e00442-20. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Hahn, W.; Bailey, B.; Hall, A.; Rodriguez, G.; Zahn, H.; Eisenberg, M.; Erickson, S. Development and Evaluation of a Sensitive Bacteriophage-Based MRSA Diagnostic Screen. Viruses 2020, 12, 631. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Garing, S.; Verma, D.; Saranathan, R.; Clute-Reinig, N.; Gadwa, J.; Peterson, C.; Hermansky, G.; Astashkina Fernandez, A.; Asare, E.; et al. Nanoluciferase Reporter Mycobacteriophage for Sensitive and Rapid Detection of Mycobacterium tuberculosis Drug Susceptibility. J. Bacteriol. 2020, 202, e00411-20. [Google Scholar] [CrossRef] [PubMed]
- Verheust, C.; Pauwels, K.; Mahillon, J.; Helinski, D.R.; Herman, P. Contained use of Bacteriophages: Risk Assessment and Biosafety Recommendations. Appl. Biosaf. 2010, 15, 32–44. [Google Scholar] [CrossRef]
- Mitsunaka, S.; Yamazaki, K.; Pramono, A.K.; Ikeuchi, M.; Kitao, T.; Ohara, N.; Kubori, T.; Nagai, H.; Ando, H. Synthetic engineering and biological containment of bacteriophages. Proc. Natl. Acad. Sci. USA 2022, 119, e2206739119. [Google Scholar] [CrossRef]
- Lemire, S.; Yehl, K.M.; Lu, T.K. Phage-Based Applications in Synthetic Biology. Annu. Rev. Virol. 2018, 5, 453–476. [Google Scholar] [CrossRef]
- Majowicz, S.E.; Musto, J.; Scallan, E.; Angulo, F.J.; Kirk, M.; O’Brien, S.J.; Jones, T.F.; Fazil, A.; Hoekstra, R.M.; International Collaboration on Enteric Disease ‘Burden of Illness’ Studies. The global burden of nontyphoidal Salmonella gastroenteritis. Clin. Infect. Dis. 2010, 50, 882–889. [Google Scholar] [CrossRef]
- Hoffmann, S.; Batz, M.B.; Morris, J.G., Jr. Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J. Food Prot. 2012, 75, 1292–1302. [Google Scholar] [CrossRef]
- Erickson, S.; Gil, J.; Stach, J.; Nguyen, M.M. Validation of PhageDx Salmonella Assay in Raw Ground Turkey and Powdered Infant Formula: AOAC Performance Tested MethodSM 121904. J. AOAC Int. 2021, 104, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Soraya Martins de Aquino, N.; Elias, S.O.; Tondo, E.C. Evaluation of PhageDX Salmonella Assay for Salmonella Detection in Hydroponic Curly Lettuce. Foods 2021, 10, 1795. [Google Scholar] [CrossRef] [PubMed]
- Soraya Martins de Aquino, N.; de Oliveira Elias, S.; Gomes, L.V.A.; Tondo, E.C. Phage-Based Assay for the Detection of Salmonella in Brazilian Poultry Products. J. Food Sci. Nutr. Res. 2021, 4, 249–258. [Google Scholar] [CrossRef]
- Akhtar, M.; Viazis, S.; Diez-Gonzalez, F. Isolation, identification and characterization of lytic, wide host range bacteriophages from waste effluents against Salmonella enterica serovars. Food Control 2014, 38, 67–74. [Google Scholar] [CrossRef]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Ruger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.B.; Revel, H.R. The genome of bacteriophage T4. Bacteriol. Rev. 1976, 40, 847–868. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.L.; Klose, T.; Arisaka, F.; Speir, J.A.; Veesler, D.; Fokine, A.; Rossmann, M.G. Role of bacteriophage T4 baseplate in regulating assembly and infection. Proc. Natl. Acad. Sci. USA 2016, 113, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Arisaka, F.; van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 355. [Google Scholar] [CrossRef]
- Yap, M.L.; Mio, K.; Leiman, P.G.; Kanamaru, S.; Arisaka, F. The baseplate wedges of bacteriophage T4 spontaneously assemble into hubless baseplate-like structure in vitro. J. Mol. Biol. 2010, 395, 349–360. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Loessner, M.J.; Rees, C.E.; Stewart, G.S.; Scherer, S. Construction of luciferase reporter bacteriophage A511::luxAB for rapid and sensitive detection of viable Listeria cells. Appl. Environ. Microbiol. 1996, 62, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Ellis, E.L.; Delbruck, M. The Growth of Bacteriophage. J. Gen. Physiol. 1939, 22, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, P.; Kim, J.H.; Ebihara, H.; Noda, T.; Neumann, G.; Feldmann, H.; Kawaoka, Y. Generation of biologically contained Ebola viruses. Proc. Natl. Acad. Sci. USA 2008, 105, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, X.; Kramer, M.F.; Zhu, J.; Brockman, M.A.; Knipe, D.M. Construction, phenotypic analysis, and immunogenicity of a UL5/UL29 double deletion mutant of herpes simplex virus 2. J. Virol. 2000, 74, 7963–7971. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; O’Neal, W.; Morral, N.; Beaudet, A.L. Development of a complementing cell line and a system for construction of adenovirus vectors with E1 and E2a deleted. J. Virol. 1996, 70, 7030–7038. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, J.S. Mutants of bacteriophage T4 unable to cause breakdown of host DNA. Proc. Natl. Acad. Sci. USA 1966, 55, 614–621. [Google Scholar] [CrossRef]
- Fry, S.E. Stimulation of recombination in phage T4 by nitrous acid-induced lesions. J. Gen. Virol. 1979, 43, 719–722. [Google Scholar] [CrossRef]
- Hadas, H.; Einav, M.; Fishov, I.; Zaritsky, A. Bacteriophage T4 development depends on the physiology of its host Escherichia coli. Microbiology 1997, 143 Pt 1, 179–185. [Google Scholar] [CrossRef]
- Strathdee, S.A.; Hatfull, G.F.; Mutalik, V.K.; Schooley, R.T. Phage therapy: From biological mechanisms to future directions. Cell 2023, 186, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Meile, S.; Baggenstos, J.; Jaggi, T.; Piffaretti, P.; Hunold, L.; Matter, C.I.; Leitner, L.; Kessler, T.M.; Loessner, M.J.; et al. Enhancing bacteriophage therapeutics through in situ production and release of heterologous antimicrobial effectors. Nat. Commun. 2023, 14, 4337. [Google Scholar] [CrossRef] [PubMed]
- Kodikara, C.P.; Crew, H.H.; Stewart, G.S. Near on-line detection of enteric bacteria using lux recombinant bacteriophage. FEMS Microbiol. Lett. 1991, 67, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Brown-Jaque, M.; Muniesa, M.; Navarro, F. Bacteriophages in clinical samples can interfere with microbiological diagnostic tools. Sci. Rep. 2016, 6, 33000. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Picazo, P.; Fernandez-Orth, D.; Brown-Jaque, M.; Miro, E.; Espinal, P.; Rodriguez-Rubio, L.; Muniesa, M.; Navarro, F. Unravelling the consequences of the bacteriophages in human samples. Sci. Rep. 2020, 10, 6737. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Pulido-Cid, M.; Chagoyen, M.; Arranz, R.; Gonzalez-Garcia, V.A.; Garcia-Doval, C.; Caston, J.R.; Valpuesta, J.M.; van Raaij, M.J.; Martin-Benito, J.; et al. Structural characterization of the bacteriophage T7 tail machinery. J. Biol. Chem. 2013, 288, 26290–26299. [Google Scholar] [CrossRef]
- Rao, V.B.; Black, L.W. Structure and assembly of bacteriophage T4 head. Virol. J. 2010, 7, 356. [Google Scholar] [CrossRef]
- Sarker, S.A.; McCallin, S.; Barretto, C.; Berger, B.; Pittet, A.C.; Sultana, S.; Krause, L.; Huq, S.; Bibiloni, R.; Bruttin, A.; et al. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 2012, 434, 222–232. [Google Scholar] [CrossRef]
- Zhang, C.; Li, W.; Liu, W.; Zou, L.; Yan, C.; Lu, K.; Ren, H. T4-like phage Bp7, a potential antimicrobial agent for controlling drug-resistant Escherichia coli in chickens. Appl. Environ. Microbiol. 2013, 79, 5559–5565. [Google Scholar] [CrossRef]
- Meile, S.; Du, J.; Staubli, S.; Grossmann, S.; Koliwer-Brandl, H.; Piffaretti, P.; Leitner, L.; Matter, C.I.; Baggenstos, J.; Hunold, L.; et al. Engineered reporter phages for detection of Escherichia coli, Enterococcus, and Klebsiella in urine. Nat. Commun. 2023, 14, 4336. [Google Scholar] [CrossRef]
- Dudek, T.; Knipe, D.M. Replication-defective viruses as vaccines and vaccine vectors. Virology 2006, 344, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Cahill, J.; Young, R. Phage Lysis: Multiple Genes for Multiple Barriers. Adv. Virus Res. 2019, 103, 33–70. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.D.; Sundarrajan, S.; Rajagopalan, S.S.; Hariharan, S.; Kempashanaiah, N.; Padmanabhan, S.; Sriram, B.; Ramachandran, J. Lysis-deficient phages as novel therapeutic agents for controlling bacterial infection. BMC Microbiol. 2011, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Lysis from without. Bacteriophage 2011, 1, 46–49. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil, J.; Paulson, J.; Zahn, H.; Brown, M.; Nguyen, M.M.; Erickson, S. Development of a Replication-Deficient Bacteriophage Reporter Lacking an Essential Baseplate Wedge Subunit. Viruses 2024, 16, 8. https://doi.org/10.3390/v16010008
Gil J, Paulson J, Zahn H, Brown M, Nguyen MM, Erickson S. Development of a Replication-Deficient Bacteriophage Reporter Lacking an Essential Baseplate Wedge Subunit. Viruses. 2024; 16(1):8. https://doi.org/10.3390/v16010008
Chicago/Turabian StyleGil, Jose, John Paulson, Henriett Zahn, Matthew Brown, Minh M. Nguyen, and Stephen Erickson. 2024. "Development of a Replication-Deficient Bacteriophage Reporter Lacking an Essential Baseplate Wedge Subunit" Viruses 16, no. 1: 8. https://doi.org/10.3390/v16010008
APA StyleGil, J., Paulson, J., Zahn, H., Brown, M., Nguyen, M. M., & Erickson, S. (2024). Development of a Replication-Deficient Bacteriophage Reporter Lacking an Essential Baseplate Wedge Subunit. Viruses, 16(1), 8. https://doi.org/10.3390/v16010008