
Comparative Evaluation of Open-Source Bioinformatics Pipelines for Full-Length Viral Genome Assembly

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.1.1. Simulated HIV-1 Quasispecies Composition (SIM)
2.1.2. Single-Genome Sequencing Data (SGS-FULL)
2.1.3. Sanger and Next-Generation Sequencing from the Same Sample (SS+NGS)
2.1.4. Next-Generation Sequencing Dataset for Runtime Benchmarking (NGS-FULL)
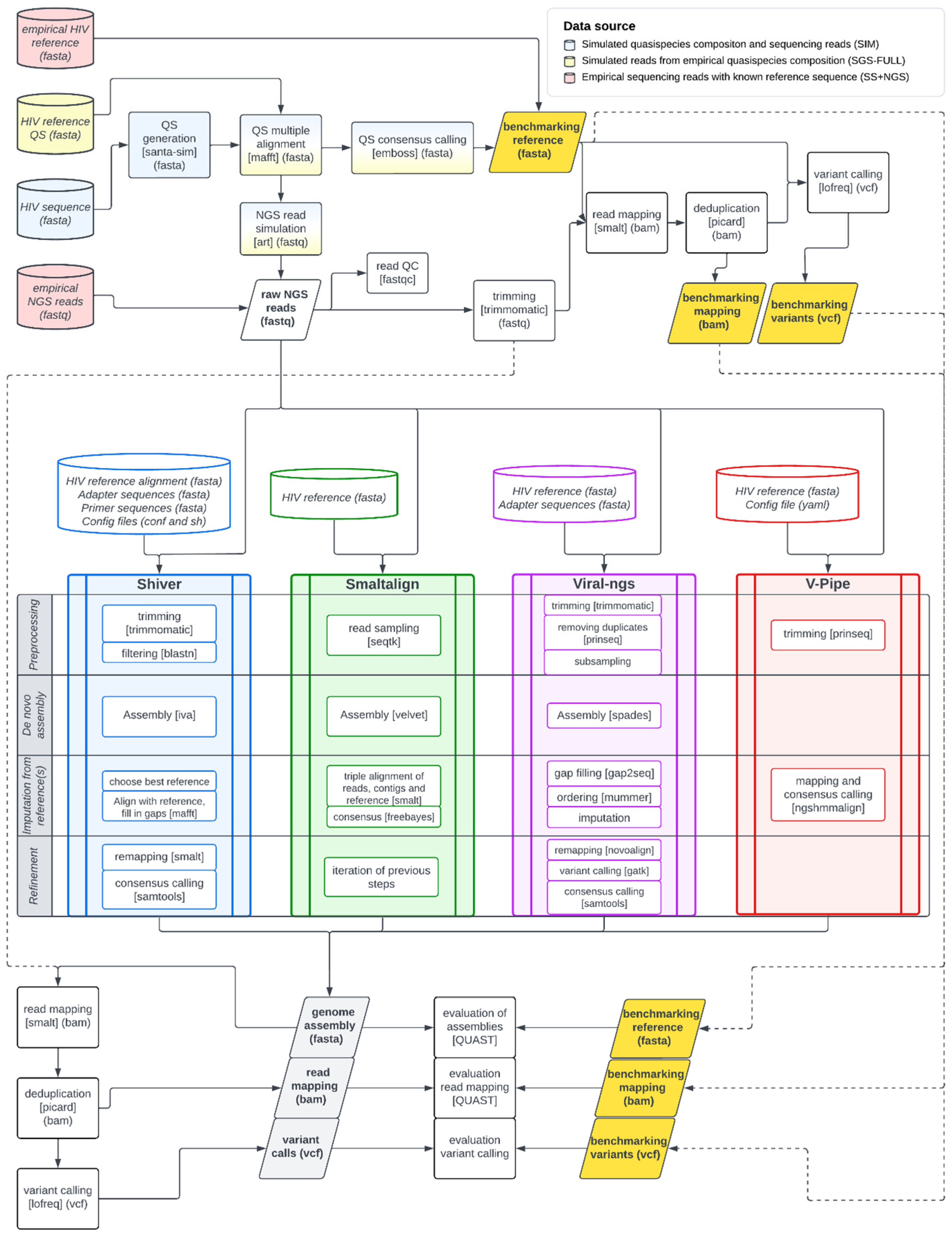
2.2. Workflow
2.2.1. Construction of Benchmarking References
2.2.2. Genome Assemblers
- The assembler must function as a data processing pipeline.
- It should be open-source.
- It should either utilize hybrid assembly methods or employ algorithms specifically designed for viral genomes.
- It must offer a command-line interface.
2.2.3. Dshiver: A Containerized Version of the Shiver Pipeline
2.2.4. Evaluation of System Requirements and the Quality of Assembled Genomes
2.2.5. Statistical Analyses
3. Results
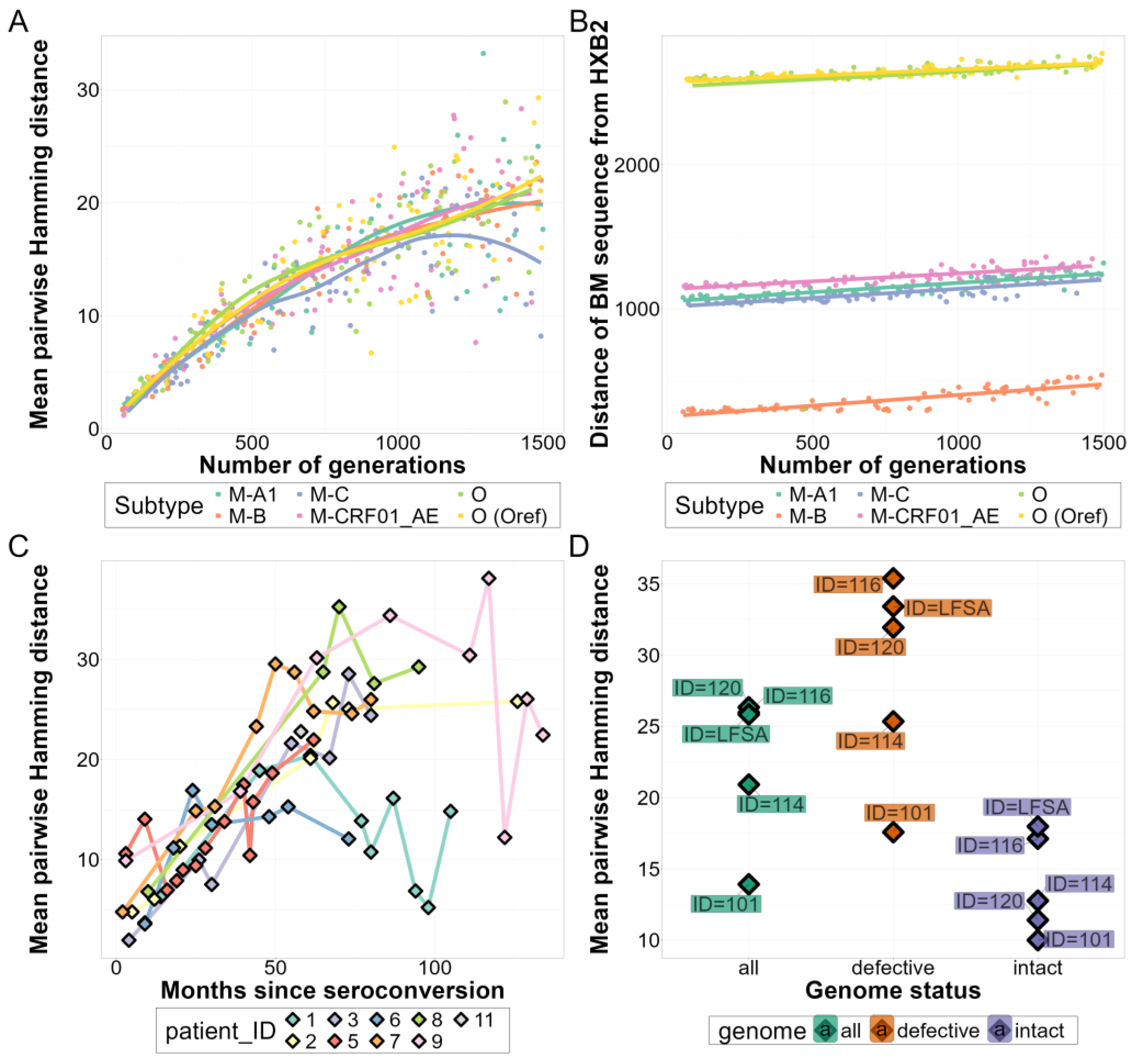
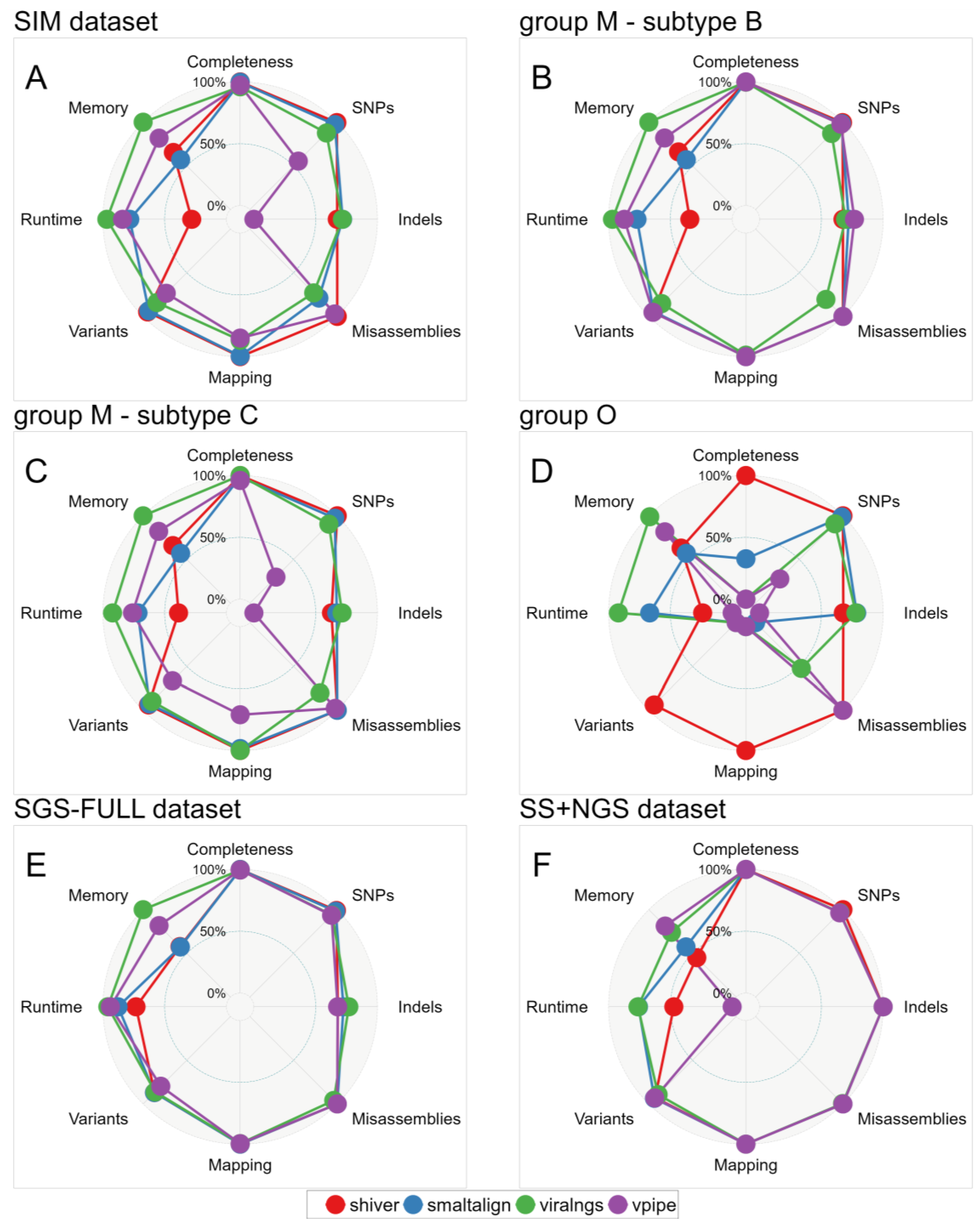
3.1. Comparison of In Vivo and In Silico Quasispecies Datasets
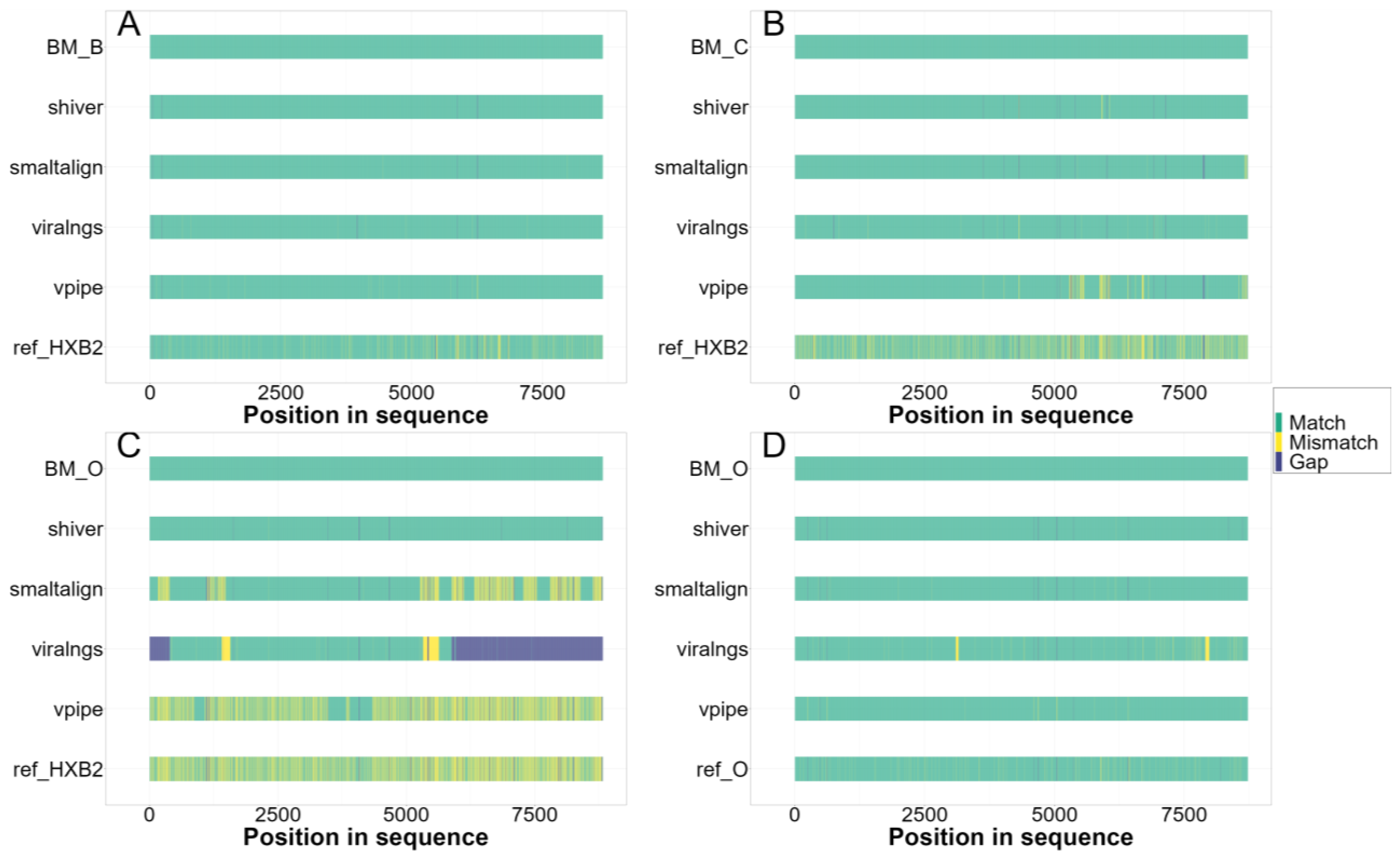
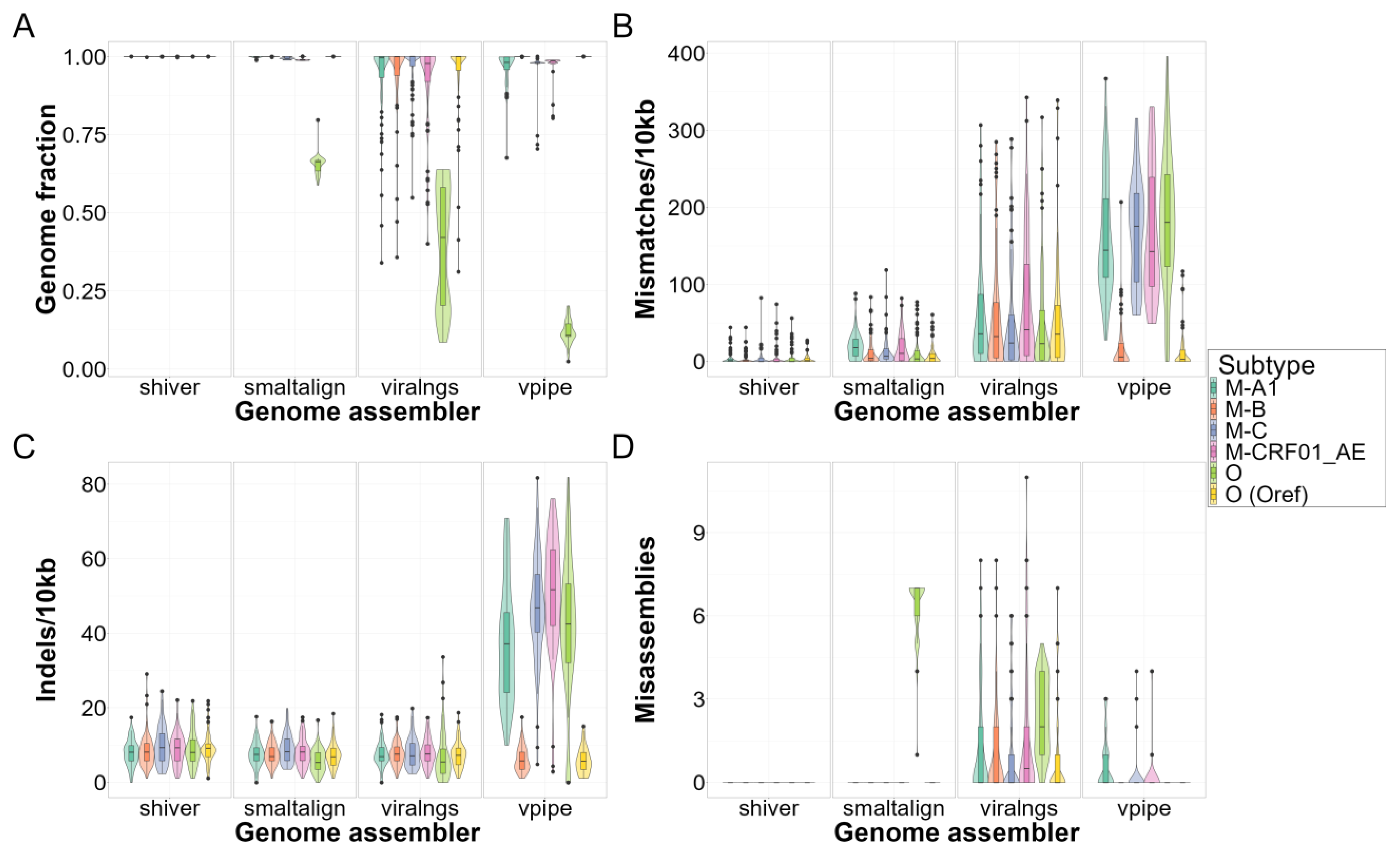
3.2. Quality of Consensus Genome Assemblies and Minority Variant Calls
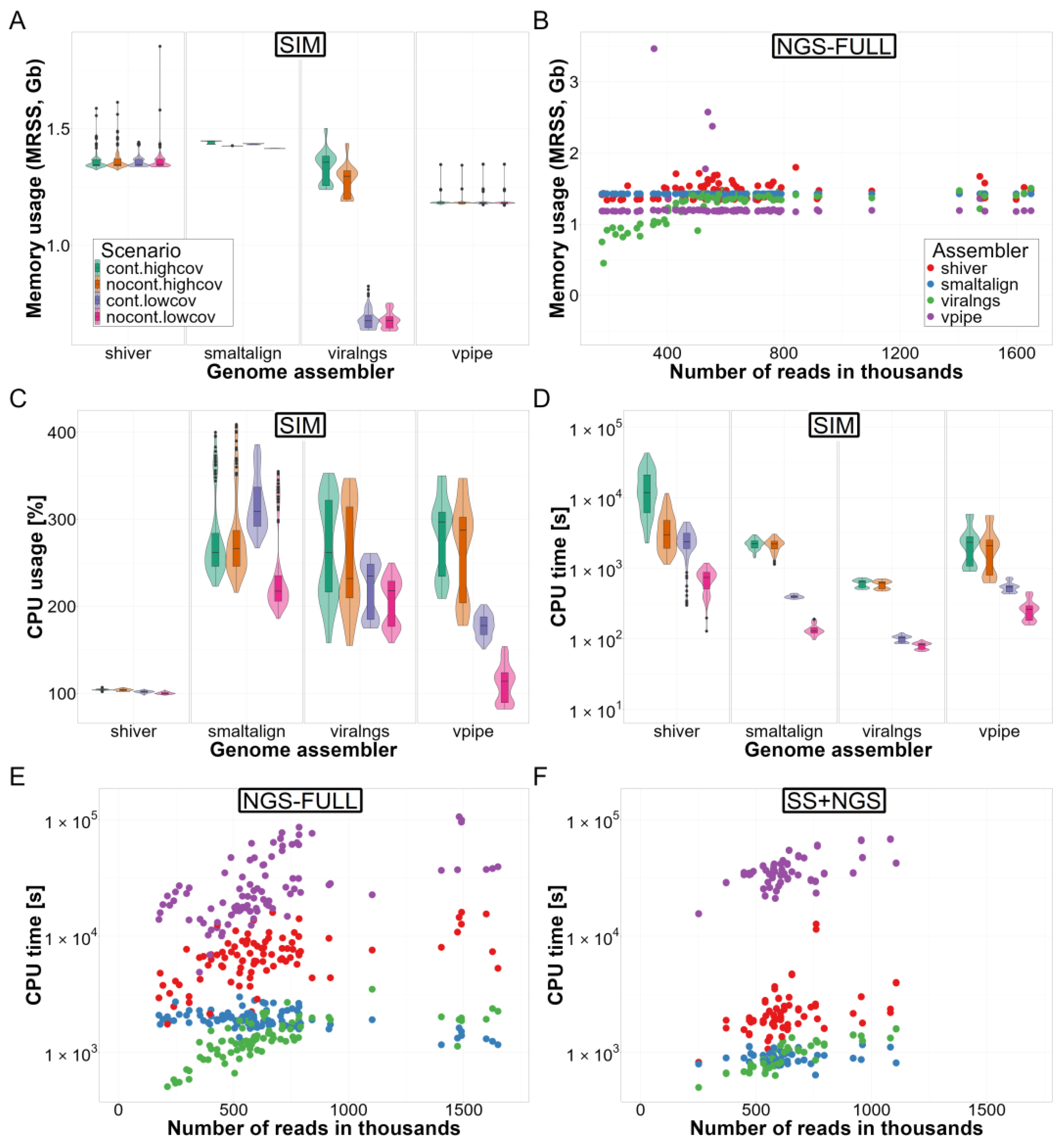
3.3. Computational Resource Use
3.4. Ease of Use
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gwinn, M.; MacCannell, D.R.; Khabbaz, R.F. Integrating Advanced Molecular Technologies into Public Health. J. Clin. Microbiol. 2017, 55, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2015, 43, D30–D35. [Google Scholar] [CrossRef] [PubMed]
- Metzner, K.J. Technologies for HIV-1 Drug Resistance Testing: Inventory and Needs. Curr. Opin. HIV AIDS 2022, 17, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Sandstrom, P.; Paredes, R.; Harrigan, P.R.; Brumme, C.J.; Avila Rios, S.; Noguera-Julian, M.; Parkin, N.; Kantor, R. Are We Ready for NGS HIV Drug Resistance Testing? The Second “Winnipeg Consensus” Symposium. Viruses 2020, 12, 586. [Google Scholar] [CrossRef]
- Gibson, R.M.; Schmotzer, C.L.; Quiñones-Mateu, M.E. Next-Generation Sequencing to Help Monitor Patients Infected with HIV: Ready for Clinical Use? Curr. Infect. Dis. Rep. 2014, 16, 401. [Google Scholar] [CrossRef]
- Dam, E.; Quercia, R.; Glass, B.; Descamps, D.; Launay, O.; Duval, X.; Kräusslich, H.-G.; Hance, A.J.; Clavel, F.; ANRS 109 Study Group. Gag Mutations Strongly Contribute to HIV-1 Resistance to Protease Inhibitors in Highly Drug-Experienced Patients besides Compensating for Fitness Loss. PLOS Pathog. 2009, 5, e1000345. [Google Scholar] [CrossRef]
- Fun, A.; Wensing, A.M.; Verheyen, J.; Nijhuis, M. Human Immunodeficiency Virus Gag and Protease: Partners in Resistance. Retrovirology 2012, 9, 63. [Google Scholar] [CrossRef]
- Yap, S.-H.; Sheen, C.-W.; Fahey, J.; Zanin, M.; Tyssen, D.; Lima, V.D.; Wynhoven, B.; Kuiper, M.; Sluis-Cremer, N.; Harrigan, P.R.; et al. N348I in the Connection Domain of HIV-1 Reverse Transcriptase Confers Zidovudine and Nevirapine Resistance. PLOS Med. 2007, 4, e335. [Google Scholar] [CrossRef]
- Malet, I.; Subra, F.; Charpentier, C.; Collin, G.; Descamps, D.; Calvez, V.; Marcelin, A.-G.; Delelis, O. Mutations Located Outside the Integrase Gene Can Confer Resistance to HIV-1 Integrase Strand Transfer Inhibitors. mBio 2017, 8, e00922-17. [Google Scholar] [CrossRef]
- Giandhari, J.; Basson, A.E.; Coovadia, A.; Kuhn, L.; Abrams, E.J.; Strehlau, R.; Morris, L.; Hunt, G.M. Genetic Changes in HIV-1 Gag-Protease Associated with Protease Inhibitor-Based Therapy Failure in Pediatric Patients. AIDS Res. Hum. Retroviruses 2015, 31, 776–782. [Google Scholar] [CrossRef]
- Hikichi, Y.; Van Duyne, R.; Pham, P.; Groebner, J.L.; Wiegand, A.; Mellors, J.W.; Kearney, M.F.; Freed, E.O. Mechanistic Analysis of the Broad Antiretroviral Resistance Conferred by HIV-1 Envelope Glycoprotein Mutations. mBio 2021, 12, e03134-20. [Google Scholar] [CrossRef] [PubMed]
- Van Duyne, R.; Kuo, L.S.; Pham, P.; Fujii, K.; Freed, E.O. Mutations in the HIV-1 Envelope Glycoprotein Can Broadly Rescue Blocks at Multiple Steps in the Virus Replication Cycle. Proc. Natl. Acad. Sci. USA 2019, 116, 9040–9049. [Google Scholar] [CrossRef]
- Monno, L.; Saracino, A.; Scudeller, L.; Punzi, G.; Brindicci, G.; Altamura, M.; Lagioia, A.; Ladisa, N.; Angarano, G. Impact of Mutations Outside the V3 Region on Coreceptor Tropism Phenotypically Assessed in Patients Infected with HIV-1 Subtype B. Antimicrob. Agents Chemother. 2011, 55, 5078–5084. [Google Scholar] [CrossRef] [PubMed]
- Vzorov, A.N.; Compans, R.W. Cytoplasmic Domain Effects on Exposure of Co-Receptor-Binding Sites of HIV-1 Env. Arch. Virol. 2016, 161, 3011–3018. [Google Scholar] [CrossRef]
- Dimonte, S.; Babakir-Mina, M.; Mercurio, F.; Di Pinto, D.; Ceccherini-Silberstein, F.; Svicher, V.; Perno, C.-F. Selected Amino Acid Changes in HIV-1 Subtype-C Gp41 Are Associated with Specific gp120V3 Signatures in the Regulation of Co-Receptor Usage. Virus Res. 2012, 168, 73–83. [Google Scholar] [CrossRef]
- Jakobsen, M.R.; Cashin, K.; Roche, M.; Sterjovski, J.; Ellett, A.; Borm, K.; Flynn, J.; Erikstrup, C.; Gouillou, M.; Gray, L.R.; et al. Longitudinal Analysis of CCR5 and CXCR4 Usage in a Cohort of Antiretroviral Therapy-Naïve Subjects with Progressive HIV-1 Subtype C Infection. PLoS ONE 2013, 8, e65950. [Google Scholar] [CrossRef]
- Thielen, A.; Sichtig, N.; Kaiser, R.; Lam, J.; Harrigan, P.R.; Lengauer, T. Improved Prediction of HIV-1 Coreceptor Usage with Sequence Information from the Second Hypervariable Loop of Gp120. J. Infect. Dis. 2010, 202, 1435–1443. [Google Scholar] [CrossRef]
- Amogne, W.; Bontell, I.; Grossmann, S.; Aderaye, G.; Lindquist, L.; Sönnerborg, A.; Neogi, U. Phylogenetic Analysis of Ethiopian HIV-1 Subtype C Near Full-Length Genomes Reveals High Intrasubtype Diversity and a Strong Geographical Cluster. AIDS Res. Hum. Retroviruses 2016, 32, 471–474. [Google Scholar] [CrossRef]
- Junqueira, D.M.; Sibisi, Z.; Wilkinson, E.; de Oliveira, T. Factors Influencing HIV-1 Phylogenetic Clustering. Curr. Opin. HIV AIDS 2019, 14, 161. [Google Scholar] [CrossRef]
- Topcu, C.; Georgiou, V.; Rodosthenous, J.H.; Kostrikis, L.G. Comparative HIV-1 Phylogenies Characterized by PR/RT, Pol and Near-Full-Length Genome Sequences. Viruses 2022, 14, 2286. [Google Scholar] [CrossRef]
- Song, H.; Giorgi, E.E.; Ganusov, V.V.; Cai, F.; Athreya, G.; Yoon, H.; Carja, O.; Hora, B.; Hraber, P.; Romero-Severson, E.; et al. Tracking HIV-1 Recombination to Resolve Its Contribution to HIV-1 Evolution in Natural Infection. Nat. Commun. 2018, 9, 1928. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; He, S.; Tian, W.; Li, D.; An, M.; Zhao, B.; Ding, H.; Xu, J.; Chu, Z.; Shang, H.; et al. First Complete-Genome Documentation of HIV-1 Intersubtype Superinfection with Transmissions of Diverse Recombinants over Time to Five Recipients. PLOS Pathog. 2021, 17, e1009258. [Google Scholar] [CrossRef] [PubMed]
- Metzner, K. HIV Whole Genome Sequencing Now—Answering Still Open Questions. J. Clin. Microbiol. 2016, 54, 834–835. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, V.; Nyandiko, W.; Vreeman, R.; DeLong, A.K.; Manne, A.; Scanlon, M.; Ngeresa, A.; Aluoch, J.; Sang, F.; Ashimosi, C.; et al. Added Value of Next Generation over Sanger Sequencing in Kenyan Youth with Extensive HIV-1 Drug Resistance. Microbiol. Spectr. 2022, 10, e03454-22. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, K.; Theys, K.; Vandamme, A.-M. HIV-1 Genotypic Drug Resistance Testing: Digging Deep, Reaching Wide? Curr. Opin. Virol. 2015, 14, 16–23. [Google Scholar] [CrossRef]
- Balakrishna, S.; Loosli, T.; Zaheri, M.; Frischknecht, P.; Huber, M.; Kusejko, K.; Yerly, S.; Leuzinger, K.; Perreau, M.; Ramette, A.; et al. Frequency Matters: Comparison of Drug Resistance Mutation Detection by Sanger and next-Generation Sequencing in HIV-1. J. Antimicrob. Chemother. 2023, 78, 656–664. [Google Scholar] [CrossRef]
- Knyazev, S.; Hughes, L.; Skums, P.; Zelikovsky, A. Epidemiological Data Analysis of Viral Quasispecies in the Next-Generation Sequencing Era. Brief. Bioinform. 2021, 22, 96–108. [Google Scholar] [CrossRef]
- Carlisle, L.A.; Turk, T.; Kusejko, K.; Metzner, K.J.; Leemann, C.; Schenkel, C.D.; Bachmann, N.; Posada, S.; Beerenwinkel, N.; Böni, J.; et al. Viral Diversity Based on Next-Generation Sequencing of HIV-1 Provides Precise Estimates of Infection Recency and Time Since Infection. J. Infect. Dis. 2019, 220, 254–265. [Google Scholar] [CrossRef]
- Leitner, T. Phylogenetics in HIV Transmission: Taking within-Host Diversity into Account. Curr. Opin. HIV AIDS 2019, 14, 181–187. [Google Scholar] [CrossRef]
- Theys, K.; Libin, P.; Pineda-Peña, A.-C.; Nowé, A.; Vandamme, A.-M.; Abecasis, A.B. The Impact of HIV-1 within-Host Evolution on Transmission Dynamics. Curr. Opin. Virol. 2018, 28, 92–101. [Google Scholar] [CrossRef]
- Wymant, C.; Hall, M.; Ratmann, O.; Bonsall, D.; Golubchik, T.; de Cesare, M.; Gall, A.; Cornelissen, M.; Fraser, C.; STOP-HCV Consortium, The Maela Pneumococcal Collaboration, and The BEEHIVE Collaboration. PHYLOSCANNER: Inferring Transmission from Within- and Between-Host Pathogen Genetic Diversity. Mol. Biol. Evol. 2018, 35, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Thrash, A.; Hoffmann, F.; Perkins, A. Toward a More Holistic Method of Genome Assembly Assessment. BMC Bioinform. 2020, 21, 249. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef] [PubMed]
- Wymant, C.; Blanquart, F.; Golubchik, T.; Gall, A.; Bakker, M.; Bezemer, D.; Croucher, N.J.; Hall, M.; Hillebregt, M.; Ong, S.H.; et al. Easy and Accurate Reconstruction of Whole HIV Genomes from Short-Read Sequence Data with Shiver. Virus Evol. 2018, 4, vey007. [Google Scholar] [CrossRef]
- Wan, Y.; Renner, D.W.; Albert, I.; Szpara, M.L. VirAmp: A Galaxy-Based Viral Genome Assembly Pipeline. GigaScience 2015, 4, 19. [Google Scholar] [CrossRef]
- Schmutz, S.; Huber, M.; Zagordi, O. SmaltAlign. 2021. Available online: https://github.com/medvir/SmaltAlign (accessed on 23 November 2024).
- Tomkins-Tinch, C.; Park, D.; Jungreis, I.; Ye, S. Viral-Ngs. Broad Institute, GitHub repository 2016. Available online: https://github.com/broadinstitute/viral-ngs (accessed on 23 November 2024).
- Jariani, A.; Warth, C.; Deforche, K.; Libin, P.; Drummond, A.J.; Rambaut, A.; Matsen IV, F.A.; Theys, K. SANTA-SIM: Simulating Viral Sequence Evolution Dynamics under Selection and Recombination. Virus Evol. 2019, 5, vez003. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Zanini, F.; Puller, V.; Brodin, J.; Albert, J.; Neher, R.A. In Vivo Mutation Rates and the Landscape of Fitness Costs of HIV-1. Virus Evol. 2017, 3, vex003. [Google Scholar] [CrossRef]
- Palmer, J.; Poon, A.F.Y. Phylogenetic Measures of Indel Rate Variation among the HIV-1 Group M Subtypes. Virus Evol. 2019, 5, vez022. [Google Scholar] [CrossRef]
- Neher, R.A.; Leitner, T. Recombination Rate and Selection Strength in HIV Intra-Patient Evolution. PLOS Comput. Biol. 2010, 6, e1000660. [Google Scholar] [CrossRef]
- Shriner, D.; Shankarappa, R.; Jensen, M.A.; Nickle, D.C.; Mittler, J.E.; Margolick, J.B.; Mullins, J.I. Influence of Random Genetic Drift on Human Immunodeficiency Virus Type 1 Env Evolution During Chronic Infection. Genetics 2004, 166, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Linchangco, G.V.; Foley, B.; Leitner, T. Updated HIV-1 Consensus Sequences Change but Stay Within Similar Distance From Worldwide Samples. Front. Microbiol. 2022, 12, 828765. [Google Scholar] [CrossRef]
- Kuiken, C.; Foley, B.; Marx, P.; Wolinsky, S.; Leitner, T.; Hahn, B.; McCutchan, F.; Korber, B.; HIV, S.C. Los Alamos HIV Sequence Database. In Theoretical Biology and Biophysics; Los Alamos National Laboratory: Los Alamos, NM, USA, 2013. [Google Scholar]
- Wang, X.Q.; Palmer, S. Single-Molecule Techniques to Quantify and Genetically Characterise Persistent HIV. Retrovirology 2018, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Vibholm, L.K.; Lorenzi, J.C.C.; Pai, J.A.; Cohen, Y.Z.; Oliveira, T.Y.; Barton, J.P.; Garcia Noceda, M.; Lu, C.-L.; Ablanedo-Terrazas, Y.; Del Rio Estrada, P.M.; et al. Characterization of Intact Proviruses in Blood and Lymph Node from HIV-Infected Individuals Undergoing Analytical Treatment Interruption. J. Virol. 2019, 93, e01920-18. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Ji, H. Next-Generation DNA Sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Áy, É.; Pocskay, Á.; Lakatos, B.; Szlávik, J.; Mezei, M.; Minárovits, J. Prevalence of Resistance Mutations Associated with Integrase Inhibitors in Therapy-Naive HIV-Positive Patients in Hungary. Acta Microbiol. Immunol. Hung. 2021, 68, 87–91. [Google Scholar] [CrossRef]
- Áy, É.; Müller, V.; Mezei, M.; Pocskay, Á.; Koroknai, A.; Müller, D.; Győri, Z.; Marschalkó, M.; Tóth, B.; Kárpáti, S.; et al. Transmitted Drug Resistance in Newly Diagnosed and Treatment-Naïve HIV Type 1-Infected Patients in Hungary. J. Glob. Antimicrob. Resist. 2020, 20, 124–130. [Google Scholar] [CrossRef]
- Mezei, M.; Ay, E.; Koroknai, A.; Tóth, R.; Balázs, A.; Bakos, A.; Gyori, Z.; Bánáti, F.; Marschalkó, M.; Kárpáti, S.; et al. Molecular Epidemiological Analysis of Env and Pol Sequences in Newly Diagnosed HIV Type 1-Infected, Untreated Patients in Hungary. AIDS Res. Hum. Retroviruses 2011, 27, 1243–1247. [Google Scholar] [CrossRef]
- Ode, H.; Matsuda, M.; Matsuoka, K.; Hachiya, A.; Hattori, J.; Kito, Y.; Yokomaku, Y.; Iwatani, Y.; Sugiura, W. Quasispecies Analyses of the HIV-1 Near-Full-Length Genome With Illumina MiSeq. Front. Microbiol. 2015, 6, 1258. [Google Scholar] [CrossRef]
- Pineda-Peña, A.-C.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gómez-López, A.; Camacho, R.J.; de Oliveira, T.; Vandamme, A.-M. Automated Subtyping of HIV-1 Genetic Sequences for Clinical and Surveillance Purposes: Performance Evaluation of the New REGA Version 3 and Seven Other Tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-Generation Sequencing Read Simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Ponstingl, H.; Ning, Z. SMALT—A New Mapper for DNA Sequencing Reads. F1000 Posters 2010, 1, 313. [Google Scholar]
- Broad Institute Picard Toolkit; Broad Institute. GitHub repository 2019. Available online: https://github.com/broadinstitute/picard (accessed on 23 November 2024).
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Park, D.J.; Dudas, G.; Wohl, S.; Goba, A.; Whitmer, S.L.M.; Andersen, K.G.; Sealfon, R.S.; Ladner, J.T.; Kugelman, J.R.; Matranga, C.B.; et al. Ebola Virus Epidemiology, Transmission, and Evolution during Seven Months in Sierra Leone. Cell 2015, 161, 1516–1526. [Google Scholar] [CrossRef]
- Posada-Céspedes, S.; Seifert, D.; Topolsky, I.; Jablonski, K.P.; Metzner, K.J.; Beerenwinkel, N. V-Pipe: A Computational Pipeline for Assessing Viral Genetic Diversity from High-Throughput Data. Bioinformatics 2021, 37, 1673–1680. [Google Scholar] [CrossRef]
- Merkel, D. Docker: Lightweight Linux Containers for Consistent Development and Deployment. Linux J. 2014, 239, 2. [Google Scholar]
- Liu, T.F.; Shafer, R.W. Web Resources for HIV Type 1 Genotypic-Resistance Test Interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Shankarappa, R.; Margolick, J.B.; Gange, S.J.; Rodrigo, A.G.; Upchurch, D.; Farzadegan, H.; Gupta, P.; Rinaldo, C.R.; Learn, G.H.; He, X.; et al. Consistent Viral Evolutionary Changes Associated with the Progression of Human Immunodeficiency Virus Type 1 Infection. J. Virol. 1999, 73, 10489–10502. [Google Scholar] [CrossRef] [PubMed]
- Ratmann, O.; Grabowski, M.K.; Hall, M.; Golubchik, T.; Wymant, C.; Abeler-Dörner, L.; Bonsall, D.; Hoppe, A.; Brown, A.L.; de Oliveira, T.; et al. Inferring HIV-1 Transmission Networks and Sources of Epidemic Spread in Africa with Deep-Sequence Phylogenetic Analysis. Nat. Commun. 2019, 10, 1411. [Google Scholar] [CrossRef] [PubMed]
- Magosi, L.E.; Zhang, Y.; Golubchik, T.; DeGruttola, V.; Tchetgen Tchetgen, E.; Novitsky, V.; Moore, J.; Bachanas, P.; Segolodi, T.; Lebelonyane, R.; et al. Deep-Sequence Phylogenetics to Quantify Patterns of HIV Transmission in the Context of a Universal Testing and Treatment Trial—BCPP/Ya Tsie Trial. eLife 2022, 11, e72657. [Google Scholar] [CrossRef]
- Monod, M.; Brizzi, A.; Galiwango, R.M.; Ssekubugu, R.; Chen, Y.; Xi, X.; Kankaka, E.N.; Ssempijja, V.; Abeler-Dörner, L.; Akullian, A.; et al. Longitudinal Population-Level HIV Epidemiologic and Genomic Surveillance Highlights Growing Gender Disparity of HIV Transmission in Uganda. Nat. Microbiol. 2024, 9, 35–54. [Google Scholar] [CrossRef]
- Chaudron, S.E.; Leemann, C.; Kusejko, K.; Nguyen, H.; Tschumi, N.; Marzel, A.; Huber, M.; Böni, J.; Perreau, M.; Klimkait, T.; et al. A Systematic Molecular Epidemiology Screen Reveals Numerous Human Immunodeficiency Virus (HIV) Type 1 Superinfections in the Swiss HIV Cohort Study. J. Infect. Dis. 2022, 226, 1256–1266. [Google Scholar] [CrossRef]
- Howison, M.; Coetzer, M.; Kantor, R. Measurement Error and Variant-Calling in Deep Illumina Sequencing of HIV. Bioinformatics 2019, 35, 2029–2035. [Google Scholar] [CrossRef]
- Alves, B.M.; Siqueira, J.D.; Prellwitz, I.M.; Botelho, O.M.; Da Hora, V.P.; Sanabani, S.; Recordon-Pinson, P.; Fleury, H.; Soares, E.A.; Soares, M.A. Estimating HIV-1 Genetic Diversity in Brazil Through Next-Generation Sequencing. Front. Microbiol. 2019, 10, 749. [Google Scholar] [CrossRef]
- Dudley, D.M.; Bailey, A.L.; Mehta, S.H.; Hughes, A.L.; Kirk, G.D.; Westergaard, R.P.; O’Connor, D.H. Cross-Clade Simultaneous HIV Drug Resistance Genotyping for Reverse Transcriptase, Protease, and Integrase Inhibitor Mutations by Illumina MiSeq. Retrovirology 2014, 11, 122. [Google Scholar] [CrossRef]
- Bennedbæk, M.; Zhukova, A.; Tang, M.-H.E.; Bennet, J.; Munderi, P.; Ruxrungtham, K.; Gisslen, M.; Worobey, M.; Lundgren, J.D.; Marvig, R.L.; et al. Phylogenetic Analysis of HIV-1 Shows Frequent Cross-Country Transmission and Local Population Expansions. Virus Evol. 2021, 7, veab055. [Google Scholar] [CrossRef]
- Struck, D.; Lawyer, G.; Ternes, A.-M.; Schmit, J.-C.; Bercoff, D.P. COMET: Adaptive Context-Based Modeling for Ultrafast HIV-1 Subtype Identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef] [PubMed]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An Automated System for Virus Identification from High-Throughput Sequencing Data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Kumar, M. Benchmarking and Assessment of Eight De Novo Genome Assemblers on Viral Next-Generation Sequencing Data, Including the SARS-CoV-2. OMICS A J. Integr. Biol. 2022, 26, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.-L.; Dhingra, A.; Fritz, A.; Götting, J.; Münch, P.C.; Steinbrück, L.; Schulz, T.F.; Ganzenmüller, T.; McHardy, A.C. Evaluating Assembly and Variant Calling Software for Strain-Resolved Analysis of Large DNA Viruses. Brief. Bioinform. 2021, 22, bbaa123. [Google Scholar] [CrossRef] [PubMed]
- Caboche, S.; Audebert, C.; Lemoine, Y.; Hot, D. Comparison of Mapping Algorithms Used in High-Throughput Sequencing: Application to Ion Torrent Data. BMC Genom. 2014, 15, 264. [Google Scholar] [CrossRef]
- Escalona, M.; Rocha, S.; Posada, D. A Comparison of Tools for the Simulation of Genomic Next-Generation Sequencing Data. Nat. Rev. Genet. 2016, 17, 459–469. [Google Scholar] [CrossRef]
- Beerenwinkel, N.; Günthard, H.F.; Roth, V.; Metzner, K.J. Challenges and Opportunities in Estimating Viral Genetic Diversity from Next-Generation Sequencing Data. Front. Microbiol. 2012, 3, 329. [Google Scholar] [CrossRef]
- Giallonardo, F.D.; Töpfer, A.; Rey, M.; Prabhakaran, S.; Duport, Y.; Leemann, C.; Schmutz, S.; Campbell, N.K.; Joos, B.; Lecca, M.R.; et al. Full-Length Haplotype Reconstruction to Infer the Structure of Heterogeneous Virus Populations. Nucleic Acids Res. 2014, 42, e115. [Google Scholar] [CrossRef]
- Kireev, D.E.; Lopatukhin, A.E.; Murzakova, A.V.; Pimkina, E.V.; Speranskaya, A.S.; Neverov, A.D.; Fedonin, G.G.; Fantin, Y.S.; Shipulin, G.A. Evaluating the Accuracy and Sensitivity of Detecting Minority HIV-1 Populations by Illumina next-Generation Sequencing. J. Virol. Methods 2018, 261, 40–45. [Google Scholar] [CrossRef]
- Alic, A.S.; Ruzafa, D.; Dopazo, J.; Blanquer, I. Objective Review of de Novo Stand-Alone Error Correction Methods for NGS Data. WIREs Comput. Mol. Sci. 2016, 6, 111–146. [Google Scholar] [CrossRef]
- Brandes, N.; Linial, M. Giant Viruses—Big Surprises. Viruses 2019, 11, 404. [Google Scholar] [CrossRef] [PubMed]
- Andreani, J.; Khalil, J.Y.B.; Baptiste, E.; Hasni, I.; Michelle, C.; Raoult, D.; Levasseur, A.; La Scola, B. Orpheovirus IHUMI-LCC2: A New Virus among the Giant Viruses. Front. Microbiol. 2018, 8, 2643. [Google Scholar] [CrossRef] [PubMed]
- Andreani, J.; Khalil, J.Y.B.; Sevvana, M.; Benamar, S.; Di Pinto, F.; Bitam, I.; Colson, P.; Klose, T.; Rossmann, M.G.; Raoult, D.; et al. Pacmanvirus, a New Giant Icosahedral Virus at the Crossroads between Asfarviridae and Faustoviruses. J. Virol. 2017, 91, e00212-17. [Google Scholar] [CrossRef] [PubMed]
- Andreani, J.; Aherfi, S.; Bou Khalil, J.Y.; Di Pinto, F.; Bitam, I.; Raoult, D.; Colson, P.; La Scola, B. Cedratvirus, a Double-Cork Structured Giant Virus, Is a Distant Relative of Pithoviruses. Viruses 2016, 8, 300. [Google Scholar] [CrossRef]
- Beaulaurier, J.; Luo, E.; Eppley, J.M.; Uyl, P.D.; Dai, X.; Burger, A.; Turner, D.J.; Pendelton, M.; Juul, S.; Harrington, E.; et al. Assembly-Free Single-Molecule Sequencing Recovers Complete Virus Genomes from Natural Microbial Communities. Genome Res. 2020, 30, 437–446. [Google Scholar] [CrossRef]
- Cao, Y.; Li, J.; Chu, X.; Liu, H.; Liu, W.; Liu, D. Nanopore Sequencing: A Rapid Solution for Infectious Disease Epidemics. Sci. China Life Sci. 2019, 62, 1101–1103. [Google Scholar] [CrossRef]
- Simmonds, P.; Bukh, J.; Combet, C.; Deléage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspé, G.; Kuiken, C.; Maertens, G.; et al. Consensus Proposals for a Unified System of Nomenclature of Hepatitis C Virus Genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A Naturally Occurring Human/Hepatitis E Recombinant Virus Predominates in Serum but Not in Faeces of a Chronic Hepatitis E Patient and Has a Growth Advantage in Cell Culture. J. Gen. Virol. 2012, 93, 526–530. [Google Scholar] [CrossRef]
- Papp, C.-P.; Biedermann, P.; Harms, D.; Wang, B.; Kebelmann, M.; Choi, M.; Helmuth, J.; Corman, V.M.; Thürmer, A.; Altmann, B.; et al. Advanced Sequencing Approaches Detected Insertions of Viral and Human Origin in the Viral Genome of Chronic Hepatitis E Virus Patients. Sci. Rep. 2022, 12, 1720. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | EM | V | DN | RB | RS | QP | CF | PC |
|---|---|---|---|---|---|---|---|---|
| shiver | -/VirtualBox/Docker 1 | v1.4.3 | + | + | + | + | + | -/- |
| SmaltAlign | Conda | v1.1.0 | + | + | - | - | - | +/- |
| viral-ngs | DNAnexus/Conda/Docker/Snakemake | v1.25.0 | + | + | - | + | - | +/+ |
| V-pipe | Snakemake (Conda) | v2.99.3 | - | + | - | + | - | +/+ |
| Metric | Reference 1 | Genome Assembler Pairwise Comparisons | Order | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SH | X | SA | SH | X | VN | SH | X | VP | SA | X | VN | SA | X | VP | VN | X | VP | |||
| Genome fraction | Matching | 0 | 7 | 1 | 6 | 2 | 0 | 0 | 8 | 0 | 5 | 3 | 0 | 0 | 8 | 0 | 0 | 3 | 5 | SA ≈ SH = VP > VN |
| Default | 16 | 0 | 0 | 15 | 1 | 0 | 16 | 0 | 0 | 9 | 7 | 0 | 16 | 0 | 0 | 4 | 12 | 0 | SH > SA > VN > VP | |
| Mismatches | Matching | 4 | 4 | 0 | 8 | 0 | 0 | 5 | 3 | 0 | 8 | 0 | 0 | 4 | 3 | 1 | 0 | 3 | 5 | SH > SA > VP > VN |
| Default | 15 | 1 | 0 | 16 | 0 | 0 | 16 | 0 | 0 | 16 | 0 | 0 | 16 | 0 | 0 | 15 | 1 | 0 | SH > SA > VN > VP | |
| Indels | Matching | 0 | 4 | 4 | 0 | 5 | 3 | 0 | 2 | 6 | 0 | 8 | 0 | 1 | 3 | 4 | 0 | 4 | 4 | VP > SA = VN > SH |
| Default | 1 | 8 | 7 | 0 | 12 | 4 | 16 | 0 | 0 | 0 | 12 | 4 | 16 | 0 | 0 | 16 | 0 | 0 | VN > SA > SH > VP | |
| Misassemblies | Matching | 0 | 8 | 0 | 4 | 4 | 0 | 0 | 8 | 0 | 5 | 3 | 0 | 0 | 8 | 0 | 0 | 3 | 5 | SA = SH = VP > VN |
| Default | 4 | 12 | 0 | 14 | 2 | 0 | 3 | 13 | 0 | 10 | 2 | 4 | 3 | 9 | 4 | 0 | 5 | 11 | SH > VP ≈ SA > VN | |
| Ns | Matching | 0 | 8 | 0 | 4 | 4 | 0 | 0 | 8 | 0 | 8 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 8 | SA = SH = VP > VN |
| Default | 0 | 16 | 0 | 10 | 6 | 0 | 0 | 16 | 0 | 16 | 0 | 0 | 0 | 16 | 0 | 0 | 0 | 16 | SH = SA = VP > VN | |
| Mapping precision | Matching | 0 | 6 | 2 | 3 | 5 | 0 | 3 | 3 | 2 | 8 | 0 | 0 | 3 | 5 | 0 | 0 | 2 | 6 | SA ≈ SH ≈ VP > VN |
| Default | 12 | 4 | 0 | 9 | 7 | 0 | 14 | 2 | 0 | 10 | 6 | 0 | 16 | 0 | 0 | 6 | 10 | 0 | SH > SA > VP > VN | |
| Variants F1 score | Matching | 0 | 4 | 4 | 3 | 5 | 0 | 0 | 4 | 4 | 8 | 0 | 0 | 1 | 4 | 3 | 0 | 0 | 8 | VP ≈ SA > SH > VN |
| Default | 11 | 5 | 0 | 10 | 6 | 0 | 14 | 2 | 0 | 10 | 6 | 0 | 16 | 0 | 0 | 8 | 8 | 0 | SH > SA > VN > VP | |
| Variants precision | Matching | 0 | 5 | 3 | 1 | 7 | 0 | 0 | 5 | 3 | 2 | 6 | 0 | 1 | 3 | 4 | 0 | 4 | 4 | VP > SA > SH ≈ VN |
| Default | 7 | 7 | 2 | 0 | 15 | 1 | 14 | 2 | 0 | 0 | 9 | 7 | 16 | 0 | 0 | 16 | 0 | 0 | VN ≈ SH > SA > VP | |
| Variants recall | Matching | 0 | 3 | 5 | 3 | 5 | 0 | 0 | 5 | 3 | 2 | 6 | 0 | 1 | 3 | 4 | 0 | 4 | 4 | VP > SA > SH > VN |
| Default | 9 | 7 | 0 | 10 | 6 | 0 | 14 | 2 | 0 | 11 | 5 | 0 | 16 | 0 | 0 | 2 | 14 | 0 | SH > SA > VN ≈ VP | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zsichla, L.; Zeeb, M.; Fazekas, D.; Áy, É.; Müller, D.; Metzner, K.J.; Kouyos, R.D.; Müller, V. Comparative Evaluation of Open-Source Bioinformatics Pipelines for Full-Length Viral Genome Assembly. Viruses 2024, 16, 1824. https://doi.org/10.3390/v16121824
Zsichla L, Zeeb M, Fazekas D, Áy É, Müller D, Metzner KJ, Kouyos RD, Müller V. Comparative Evaluation of Open-Source Bioinformatics Pipelines for Full-Length Viral Genome Assembly. Viruses. 2024; 16(12):1824. https://doi.org/10.3390/v16121824
Chicago/Turabian StyleZsichla, Levente, Marius Zeeb, Dávid Fazekas, Éva Áy, Dalma Müller, Karin J. Metzner, Roger D. Kouyos, and Viktor Müller. 2024. "Comparative Evaluation of Open-Source Bioinformatics Pipelines for Full-Length Viral Genome Assembly" Viruses 16, no. 12: 1824. https://doi.org/10.3390/v16121824
APA StyleZsichla, L., Zeeb, M., Fazekas, D., Áy, É., Müller, D., Metzner, K. J., Kouyos, R. D., & Müller, V. (2024). Comparative Evaluation of Open-Source Bioinformatics Pipelines for Full-Length Viral Genome Assembly. Viruses, 16(12), 1824. https://doi.org/10.3390/v16121824