
Optimizing the Multimerization Properties of Quinoline-Based Allosteric HIV-1 Integrase Inhibitors

Abstract
:1. Introduction
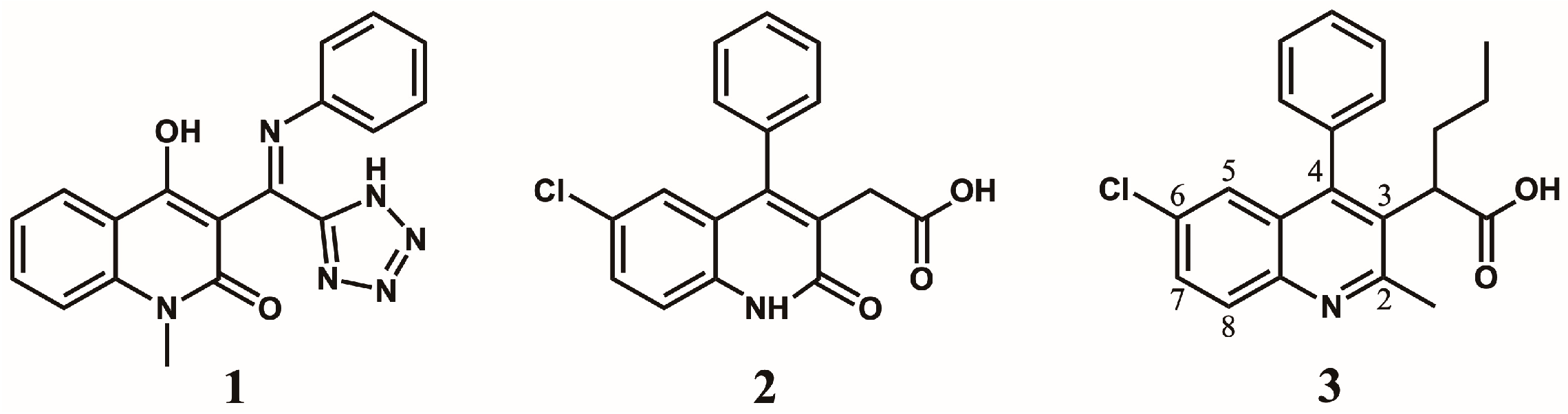
2. Discovery and Initial Optimization of the Quinoline-Based ALLINIs
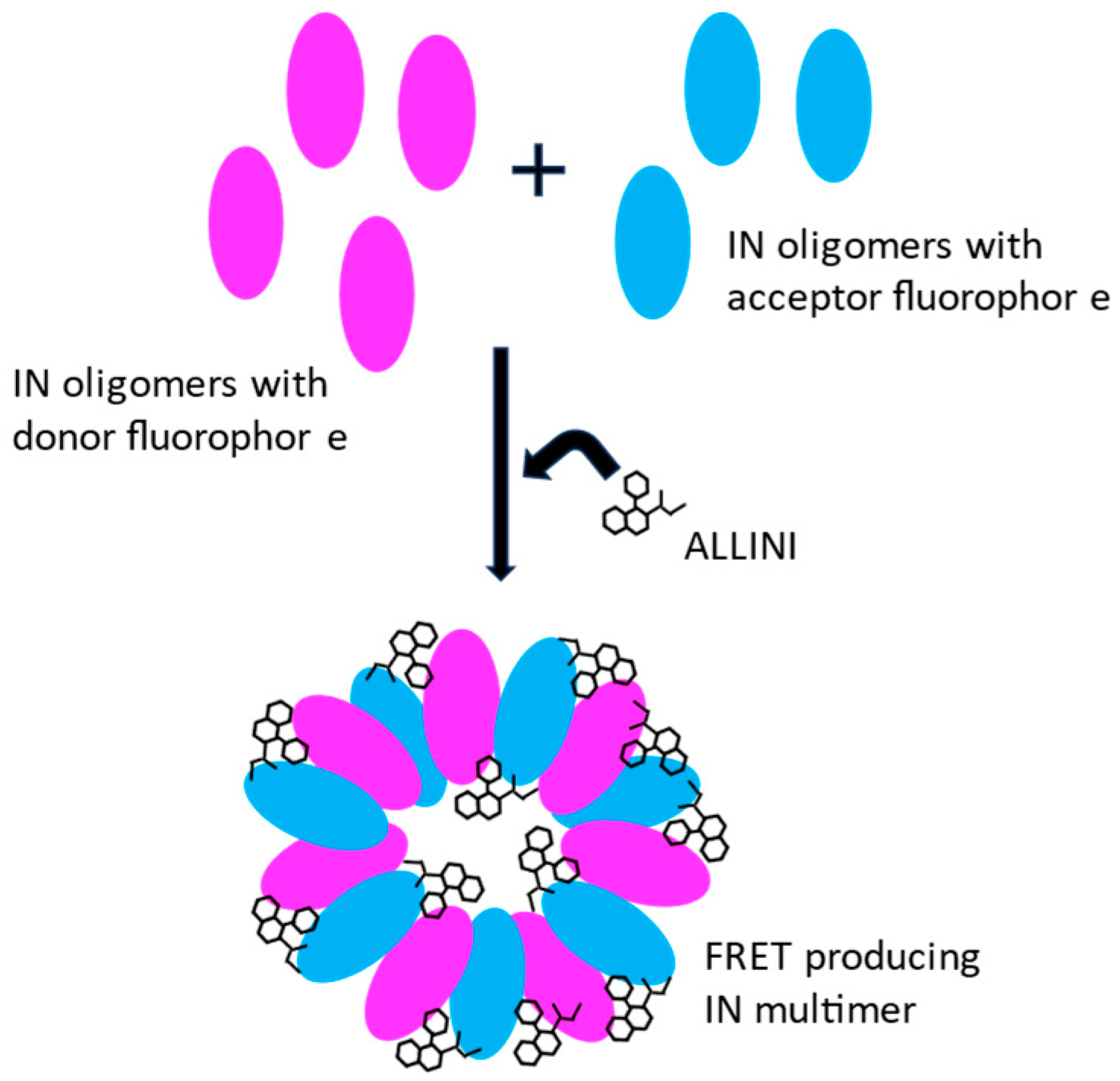
3. Integrase Multimerization by ALLINIs
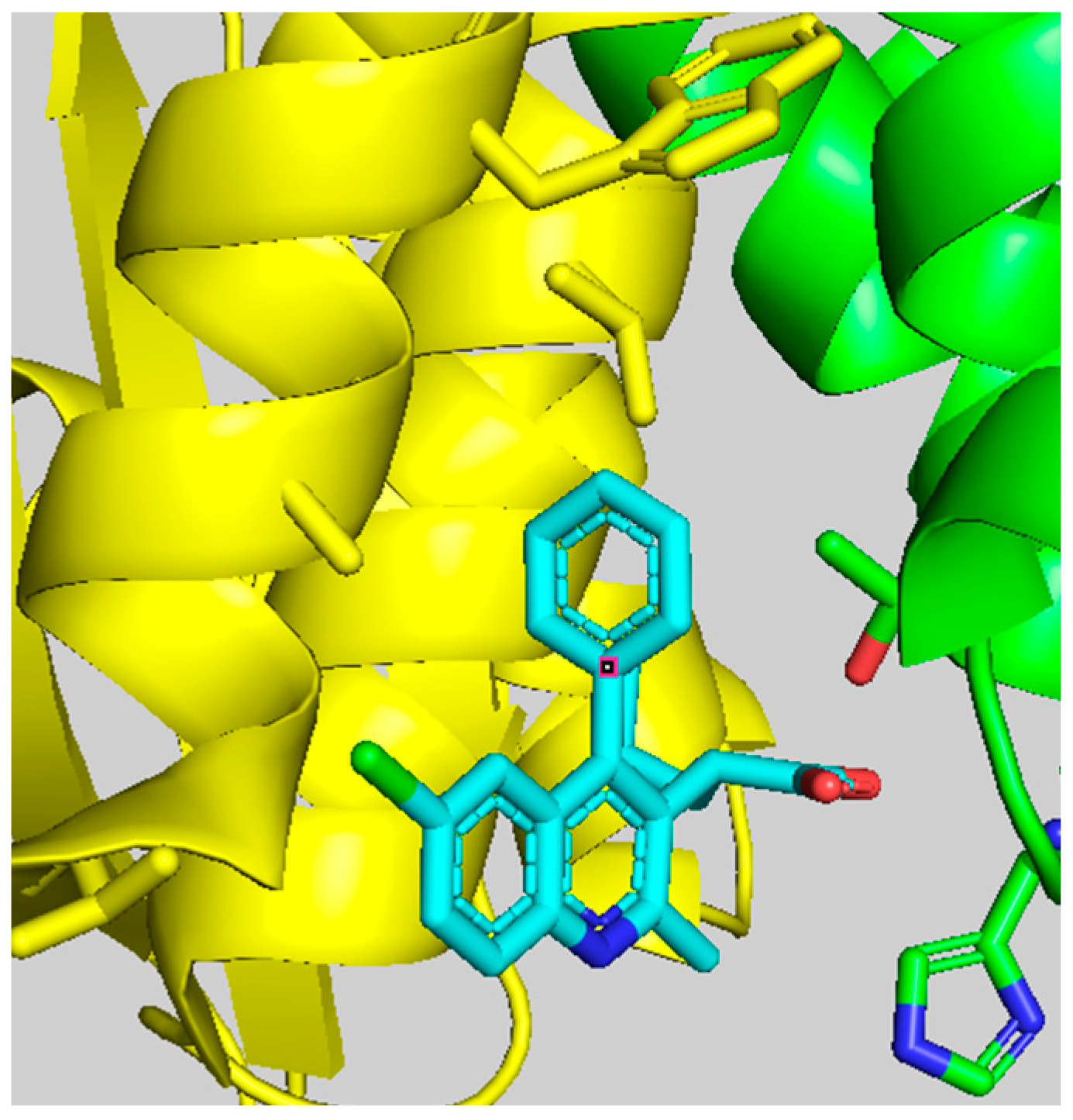
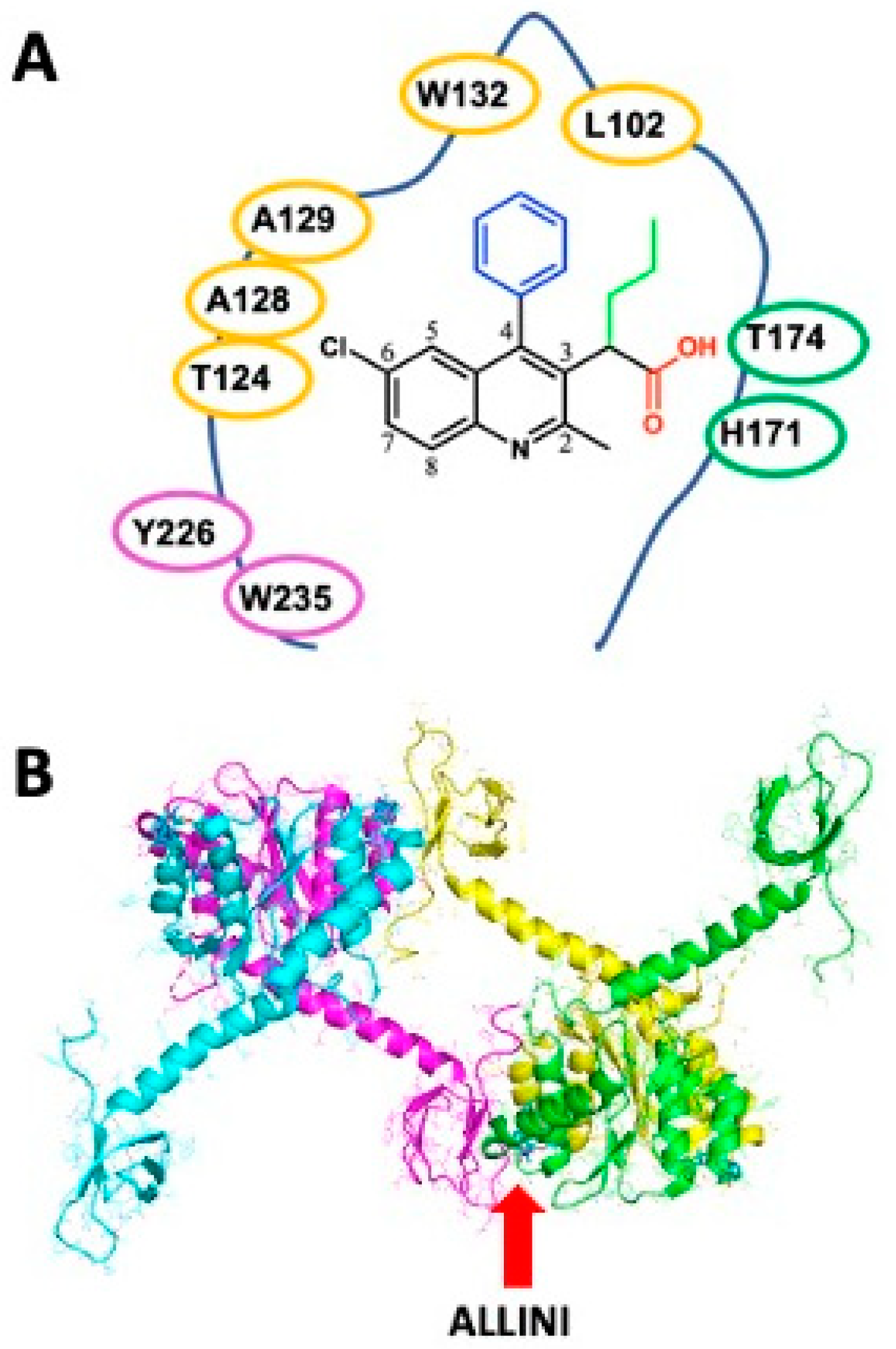
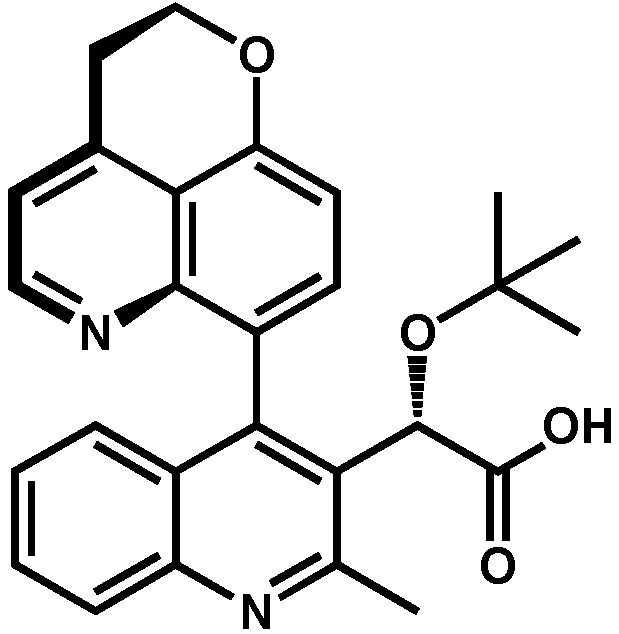
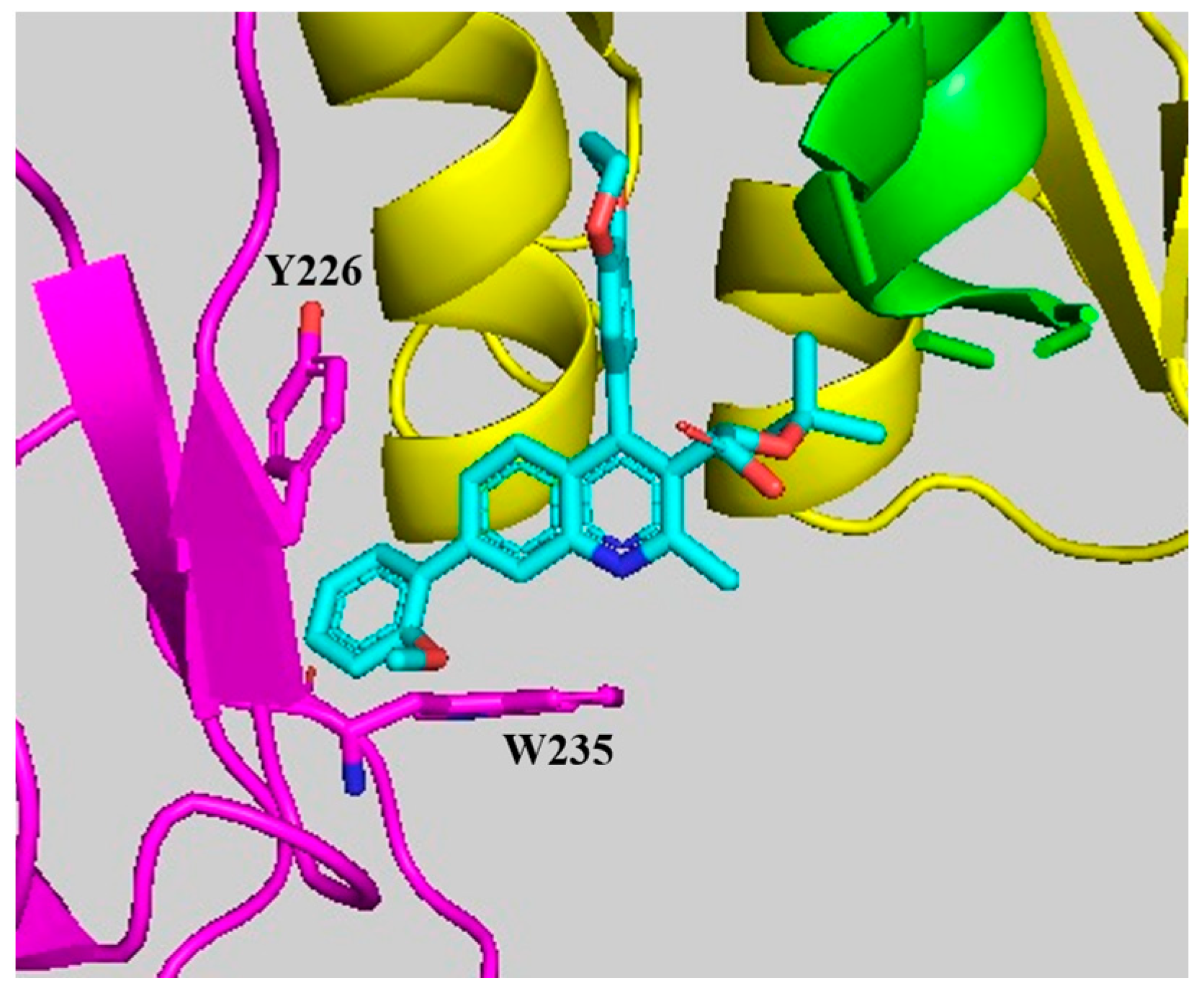
4. Binding Features of Quinoline-Based ALLINIs
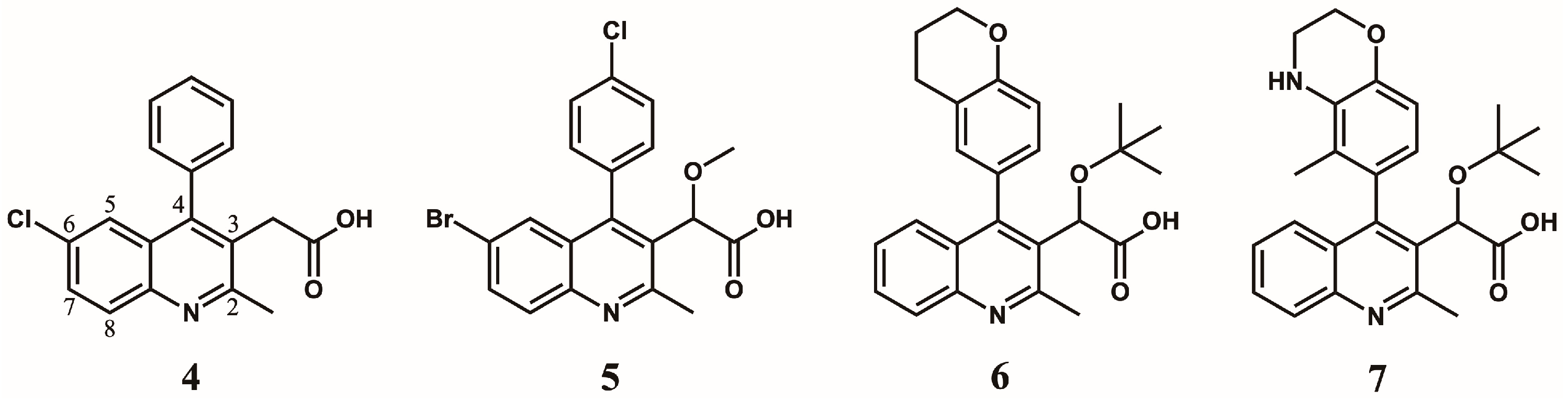
5. Optimization of the Quinoline-Based ALLINIs
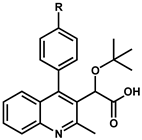
5.1. Optimization of Position 4
5.2. Optimization of Position 6
5.3. Optimization of Position 7
5.4. Optimization of Position 8
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R., Jr.; Hughes, S.H. Integrase Strand Transfer Inhibitors Are Effective Anti-HIV Drugs. Viruses 2021, 13, 205. [Google Scholar] [CrossRef]
- Quashie, P.K.; Mesplede, T.; Wainberg, M.A. Evolution of HIV integrase resistance mutations. Curr. Opin. Infect Dis. 2013, 26, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Tu, N.Q.; Delelis, O. Different Pathways Conferring Integrase Strand-Transfer Inhibitors Resistance. Viruses 2022, 14, 2591. [Google Scholar] [CrossRef]
- Chiu, T.K.; Davies, D.R. Structure and function of HIV-1 integrase. Curr. Top Med. Chem. 2004, 4, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. Retroviral Integrase Structure and DNA Recombination Mechanism. Microbiol. Spectr. 2014, 2, 1011–1033. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Engelman, A. Retroviral integrase proteins and HIV-1 DNA integration. J. Biol. Chem. 2012, 287, 40858–40866. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.N.; Hare, S. Structural insights into the retroviral DNA integration apparatus. Curr. Opin. Struct. Biol. 2011, 21, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Krishnan, L.; Cherepanov, P.; Engelman, A. Structural biology of retroviral DNA integration. Virology 2011, 411, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef]
- Passos, D.O.; Li, M.; Yang, R.; Rebensburg, S.V.; Ghirlando, R.; Jeon, Y.; Shkriabai, N.; Kvaratskhelia, M.; Craigie, R.; Lyumkis, D. Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science 2017, 355, 89–92. [Google Scholar] [CrossRef]
- Busschots, K.; Vercammen, J.; Emiliani, S.; Benarous, R.; Engelborghs, Y.; Christ, F.; Debyser, Z. The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J. Biol. Chem. 2005, 280, 17841–17847. [Google Scholar] [CrossRef]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef]
- Shun, M.C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Ambrosio, A.L.; Rahman, S.; Ellenberger, T.; Engelman, A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. USA 2005, 102, 17308–17313. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Saenz, D.T.; Meehan, A.; Wongthida, P.; Peretz, M.; Walker, W.H.; Teo, W.; Poeschla, E.M. An essential role for LEDGF/p75 in HIV integration. Science 2006, 314, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Sun, Z.Y.; Rahman, S.; Maertens, G.; Wagner, G.; Engelman, A. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat. Struct. Mol. Biol. 2005, 12, 526–532. [Google Scholar] [CrossRef]
- Kessl, J.J.; Jena, N.; Koh, Y.; Taskent-Sezgin, H.; Slaughter, A.; Feng, L.; de Silva, S.; Wu, L.; Le Grice, S.F.; Engelman, A.; et al. Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. [Google Scholar] [CrossRef]
- Feng, L.; Sharma, A.; Slaughter, A.; Jena, N.; Koh, Y.; Shkriabai, N.; Larue, R.C.; Patel, P.A.; Mitsuya, H.; Kessl, J.J.; et al. The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 2013, 288, 15813–15820. [Google Scholar] [CrossRef]
- Sharma, A.; Slaughter, A.; Jena, N.; Feng, L.; Kessl, J.J.; Fadel, H.J.; Malani, N.; Male, F.; Wu, L.; Poeschla, E.; et al. A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS Pathog. 2014, 10, e1004171. [Google Scholar] [CrossRef]
- Patel, P.A.; Kvaratskhelia, N.; Mansour, Y.; Antwi, J.; Feng, L.; Koneru, P.; Kobe, M.J.; Jena, N.; Shi, G.; Mohamed, M.S.; et al. Indole-based allosteric inhibitors of HIV-1 integrase. Bioorg. Med. Chem. Lett. 2016, 26, 4748–4752. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, N.G.; Hart, A.P.; Hume, J.D.; Sun, J.; McNeely, K.A.; Lama, C.; Pigza, J.A.; Donahue, M.G.; Kessl, J.J. Synthesis and Evaluation of Aryl Quinolines as HIV-1 Integrase Multimerization Inhibitors. ACS Med. Chem. Lett. 2018, 9, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Tsiang, M.; Jones, G.S.; Niedziela-Majka, A.; Kan, E.; Lansdon, E.B.; Huang, W.; Hung, M.; Samuel, D.; Novikov, N.; Xu, Y.; et al. New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J. Biol. Chem. 2012, 287, 21189–21203. [Google Scholar] [CrossRef]
- Bonnard, D.; Le Rouzic, E.; Eiler, S.; Amadori, C.; Orlov, I.; Bruneau, J.M.; Brias, J.; Barbion, J.; Chevreuil, F.; Spehner, D.; et al. Structure-function analyses unravel distinct effects of allosteric inhibitors of HIV-1 integrase on viral maturation and integration. J. Biol. Chem. 2018, 293, 6172–6186. [Google Scholar] [CrossRef]
- Fenwick, C. Identification of BI-C, a novel HIV-1 non-catalytic site integrase inhibitor. In Proceedings of the 18th Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 9 February 2011. [Google Scholar]
- Sierecki, E.; Giles, N.; Polinkovsky, M.; Moustaqil, M.; Alexandrov, K.; Gambin, Y. A cell-free approach to accelerate the study of protein-protein interactions in vitro. Interface Focus 2013, 3, 20130018. [Google Scholar] [CrossRef]
- Han, Y.S.; Quashie, P.; Mesplede, T.; Xu, H.; Mekhssian, K.; Fenwick, C.; Wainberg, M.A. A high-throughput assay for HIV-1 integrase 3’-processing activity using time-resolved fluorescence. J. Virol. Methods 2012, 184, 34–40. [Google Scholar] [CrossRef]
- Fenwick, C.W.; Tremblay, S.; Wardrop, E.; Bethell, R.; Coulomb, R.; Elston, R.; Faucher, A.M.; Mason, S.; Simoneau, B.; Tsantrizos, Y.; et al. Resistance studies with HIV-1 non-catalytic site integrase inhibitors. Antivir. Ther. 2011, 16, A9. [Google Scholar]
- Kessl, J.J.; Sharma, A.; Kvaratskhelia, M. Methods for the Analyses of Inhibitor-Induced Aberrant Multimerization of HIV-1 Integrase. Methods Mol. Biol. 2016, 1354, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Desimmie, B.A.; Schrijvers, R.; Demeulemeester, J.; Borrenberghs, D.; Weydert, C.; Thys, W.; Vets, S.; Van Remoortel, B.; Hofkens, J.; De Rijck, J.; et al. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 2013, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, M.; Yant, S.R.; Tsai, L.; O’Sullivan, C.; Bam, R.A.; Tsai, A.; Niedziela-Majka, A.; Stray, K.M.; Sakowicz, R.; Cihlar, T. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS ONE 2013, 8, e74163. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, E.; Bonnard, D.; Chasset, S.; Bruneau, J.M.; Chevreuil, F.; Le Strat, F.; Nguyen, J.; Beauvoir, R.; Amadori, C.; Brias, J.; et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 2013, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Kutluay, S.B.; Townsend, D.; Rebensburg, S.; Slaughter, A.; Larue, R.C.; Shkriabai, N.; Bakouche, N.; Fuchs, J.R.; Bieniasz, P.D.; et al. HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 2016, 166, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Hoyte, A.; Mansour, Y.E.; Mohamed, M.S.; Fuchs, J.R.; Engelman, A.N.; Kvaratskhelia, M.; Levy, R. Allosteric HIV-1 integrase inhibitors promote aberrant protein multimerization by directly mediating inter-subunit interactions: Structural and thermodynamic modeling studies. Protein Sci. 2016, 25, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Patel, K.; Hume, J.; Pigza, J.A.; Donahue, M.G.; Kessl, J.J. Optimized binding of substituted quinoline ALLINIs within the HIV-1 integrase oligomer. J. Biol. Chem. 2021, 296, 100363. [Google Scholar] [CrossRef]
- Dinh, L.P.; Sun, J.; Glenn, C.D.; Patel, K.; Pigza, J.A.; Donahue, M.G.; Yet, L.; Kessl, J.J. Multi-Substituted Quinolines as HIV-1 Integrase Allosteric Inhibitors. Viruses 2022, 14, 1466. [Google Scholar] [CrossRef]
- Gupta, K.; Brady, T.; Dyer, B.M.; Malani, N.; Hwang, Y.; Male, F.; Nolte, R.T.; Wang, L.P.; Velthuisen, E.; Jeffrey, J.; et al. Allosteric Inhibition of Human Immunodeficiency Virus Integrase. J. Biol. Chem. 2014, 289, 20477–20488. [Google Scholar] [CrossRef]
- Gupta, K.; Turkki, V.; Sherrill-Mix, S.; Hwang, Y.; Eilers, G.; Taylor, L.; McDanal, C.; Wang, P.; Temelkoff, D.; Nolte, R.T.; et al. Structural Basis for Inhibitor-Induced Aggregation of HIV Integrase. PLoS Biol. 2016, 14, e1002584. [Google Scholar] [CrossRef]
- Gupta, K.; Allen, A.; Giraldo, C.; Eilers, G.; Sharp, R.; Hwang, Y.; Murali, H.; Cruz, K.; Janmey, P.; Bushman, F.; et al. Allosteric HIV Integrase Inhibitors Promote Formation of Inactive Branched Polymers via Homomeric Carboxy-Terminal Domain Interactions. Structure 2021, 29, 213–225. [Google Scholar] [CrossRef]
- Eilers, G.; Gupta, K.; Allen, A.; Montermoso, S.; Murali, H.; Sharp, R.; Hwang, Y.; Bushman, F.D.; Van Duyne, G. Structure of a HIV-1 IN-Allosteric inhibitor complex at 2.93 Å resolution: Routes to inhibitor optimization. PLoS Pathog. 2023, 19, e1011097. [Google Scholar] [CrossRef]
- Koneru, P.C.; Francis, A.C.; Deng, N.; Rebensburg, S.V.; Hoyte, A.C.; Lindenberger, J.; Adu-Ampratwum, D.; Larue, R.C.; Wempe, M.F.; Engelman, A.N.; et al. HIV-1 integrase tetramers are the antiviral target of pyridine-based allosteric integrase inhibitors. eLife 2019, 8, e46344. [Google Scholar] [CrossRef]
- Shkriabai, N.; Dharmarajan, V.; Slaughter, A.; Kessl, J.J.; Larue, R.C.; Feng, L.; Fuchs, J.R.; Griffin, P.R.; Kvaratskhelia, M. A critical role of the C-terminal segment for allosteric inhibitor-induced aberrant multimerization of HIV-1 integrase. J. Biol. Chem. 2014, 289, 26430–26440. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.N.; Inoue, Y.; Nakanishi, I.; Kitaura, K. Cl-pi interactions in protein-ligand complexes. Protein Sci. 2008, 17, 1129–1137. [Google Scholar] [CrossRef]
- Fader, L.D.; Malenfant, E.; Parisien, M.; Carson, R.; Bilodeau, F.; Landry, S.; Pesant, M.; Brochu, C.; Morin, S.; Chabot, C.; et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 2014, 5, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Fandrick, K.R.; Li, W.; Zhang, Y.; Tang, W.; Gao, J.; Rodriguez, S.; Patel, N.D.; Reeves, D.C.; Wu, J.P.; Sanyal, S.; et al. Concise and Practical Asymmetric Synthesis of a Challenging Atropisomeric HIV Integrase Inhibitor. Angew. Chem. Int. Ed. Engl. 2015, 54, 7144–7148. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, C.; Amad, M.; Bailey, M.D.; Bethell, R.; Bos, M.; Bonneau, P.; Cordingley, M.; Coulombe, R.; Duan, J.; Edwards, P.; et al. Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3233–3244. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compound | R | EC50 (µM) a,b | Compound | R | EC50 (µM) a,b |
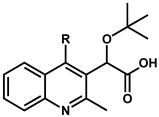
| 8 | H | 1.32 ± 0.53 | 14 | OCF3 | 0.35 ± 0.09 |
| 9 | Cl | 0.10 ± 0.02 | 15 | SCH3 | 0.49 ± 0.01 |
| 10 | F | 0.49 ± 0.04 | 16 | CN | 2.97 ± 0.81 |
| 11 | CH3 | 0.24 ± 0.11 | 17 | COCH3 | 1.39 ± 0.48 |
| 12 | CF3 | 0.72 ± 0.06 | 18 | NHCOCH3 | 8.41 ± 0.73 |
| 13 | OCH3 | 0.23 ± 0.04 | 19 | Phenyl | no activity |
 | ||||||
|---|---|---|---|---|---|---|
| R-para | R-meta | R-ortho | ||||
| R group | Compound (R-para) | EC50 (µM) a,b | Compound (R-meta) | EC50 (µM) a,b | Compound (R-ortho) | EC50 (µM) a,b |
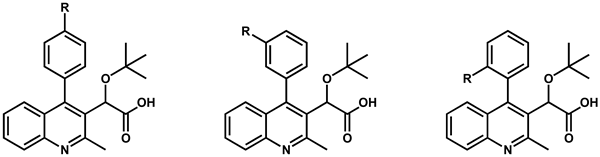
| Cl | 9 | 0.10 ± 0.02 | 20 | 3.79 ± 0.09 | 26 | 0.26 ± 0.04 |
| F | 10 | 0.49 ± 0.04 | 21 | 2.11 ± 0.59 | 27 | 0.58 ± 0.07 |
| CH3 | 11 | 0.24 ± 0.11 | 22 | 0.95 ± 0.29 | 28 | 0.51 ± 0.05 |
| OCH3 | 13 | 0.23 ± 0.04 | 23 | 1.38 ± 0.37 | 29 | 8.39 ± 0.92 |
| SCH3 | 15 | 0.49 ± 0.01 | 24 | 1.00 ± 0.14 | 30 | 4.51 ± 0.71 |
| CN | 16 | 2.97 ± 0.81 | 25 | no activity | 31 | 11.14 ± 0.85 |
 | |||||
|---|---|---|---|---|---|
| Compound | R | EC50 (µM) a,b | Compound | R | EC50 (µM) a,b |
| 32 |  | 0.25 c | 6 |  | 0.03 ± 0.01 d |
| 33 |  | 3.70 ± 0.90 | 36 | 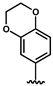 | 0.08 ± 0.01 |
| 34 | 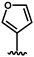 | 2.21 ± 0.56 | 37 | 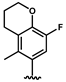 | 0.02 c |
| 35 | 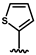 | no activity | 38 |  | no activity |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R | EC50 (µM) a,b | Compound | R | EC50 (µM) a,b | Compound | R | EC50 (µM) a,b |
| 39 | Br | 0.09 ± 0.01 0.07 ± 0.01 c | 46 |  | 1.28 ± 0.01 | 53 |  | no activity |
| 40 | I | 0.20 ± 0.06 | 47 |  | 1.30 ± 0.37 | 54 |  | 1.08 ± 0.17 |
| 41 | NH2 | 0.19 ± 0.05 | 48 |  | 1.20 ± 0.14 | 55 | 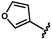 | no activity |
| 42 | 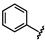 | 1.17 ± 0.14 | 49 | 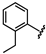 | no activity | 56 | 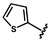 | 1.53 ± 0.13 |
| 43 | 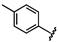 | 1.59 ± 0.04 | 50 | 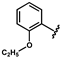 | no activity | 57 |  | 1.53 ± 0.33 |
| 44 | 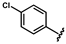 | 1.93 ± 0.01 | 51 | 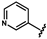 | 0.57 ± 0.29 | 58 |  | no activity |
| 45 |  | 1.29 ± 0.01 | 52 |  | 0.97 ± 0.40 | 59 |  | no activity |
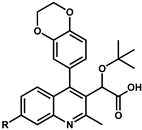 | |||||
|---|---|---|---|---|---|
| Compound | R | EC50 (µM) a,b | Compound | R | EC50 (µM) a,b |
| 36 | H | 0.08 ± 0.01 | 64 |  | 0.12 ± 0.01 |
| 60 |  | 0.07 ± 0.01 | 65 | 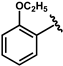 | 0.06 ± 0.01 |
| 61 | 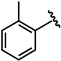 | 0.06 ± 0.01 | 66 |  | 0.04 ± 0.01 |
| 62 | 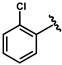 | 0.11 ± 0.01 | 67 |  | 0.52 ± 0.01 |
| 63 | 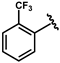 | 0.14 ± 0.01 | 68 | 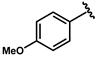 | 0.04 ± 0.01 |
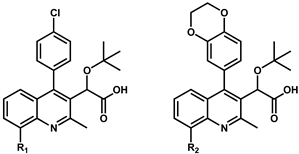 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | EC50 (µM) a,b | Compound | R1 | EC50 (µM) a,b | Compound | R2 | EC50 (µM) a,b |
| 9 | H | 0.10 ± 0.02 | 71 | 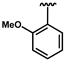 | 0.26 ± 0.04 | 36 | H | 0.08 ± 0.01 |
| 69 | Br | 0.09 ± 0.01 | 72 | 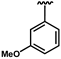 | 0.24 ± 0.01 | 74 | Cl | 0.08 ± 0.01 |
| 70 | 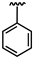 | 0.28 ± 0.03 | 73 | 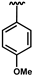 | 0.35 ± 0.08 | 75 | Br | 0.05 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Kessl, J.J. Optimizing the Multimerization Properties of Quinoline-Based Allosteric HIV-1 Integrase Inhibitors. Viruses 2024, 16, 200. https://doi.org/10.3390/v16020200
Sun J, Kessl JJ. Optimizing the Multimerization Properties of Quinoline-Based Allosteric HIV-1 Integrase Inhibitors. Viruses. 2024; 16(2):200. https://doi.org/10.3390/v16020200
Chicago/Turabian StyleSun, Jian, and Jacques J. Kessl. 2024. "Optimizing the Multimerization Properties of Quinoline-Based Allosteric HIV-1 Integrase Inhibitors" Viruses 16, no. 2: 200. https://doi.org/10.3390/v16020200