
Abundant Intra-Subtype Reassortment Revealed in H13N8 Influenza Viruses
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Virus Isolation and Detection
2.3. Virus Sequencing
2.4. Preparation of Concatenated Sequences and Alignment
2.5. Reassortment Analysis
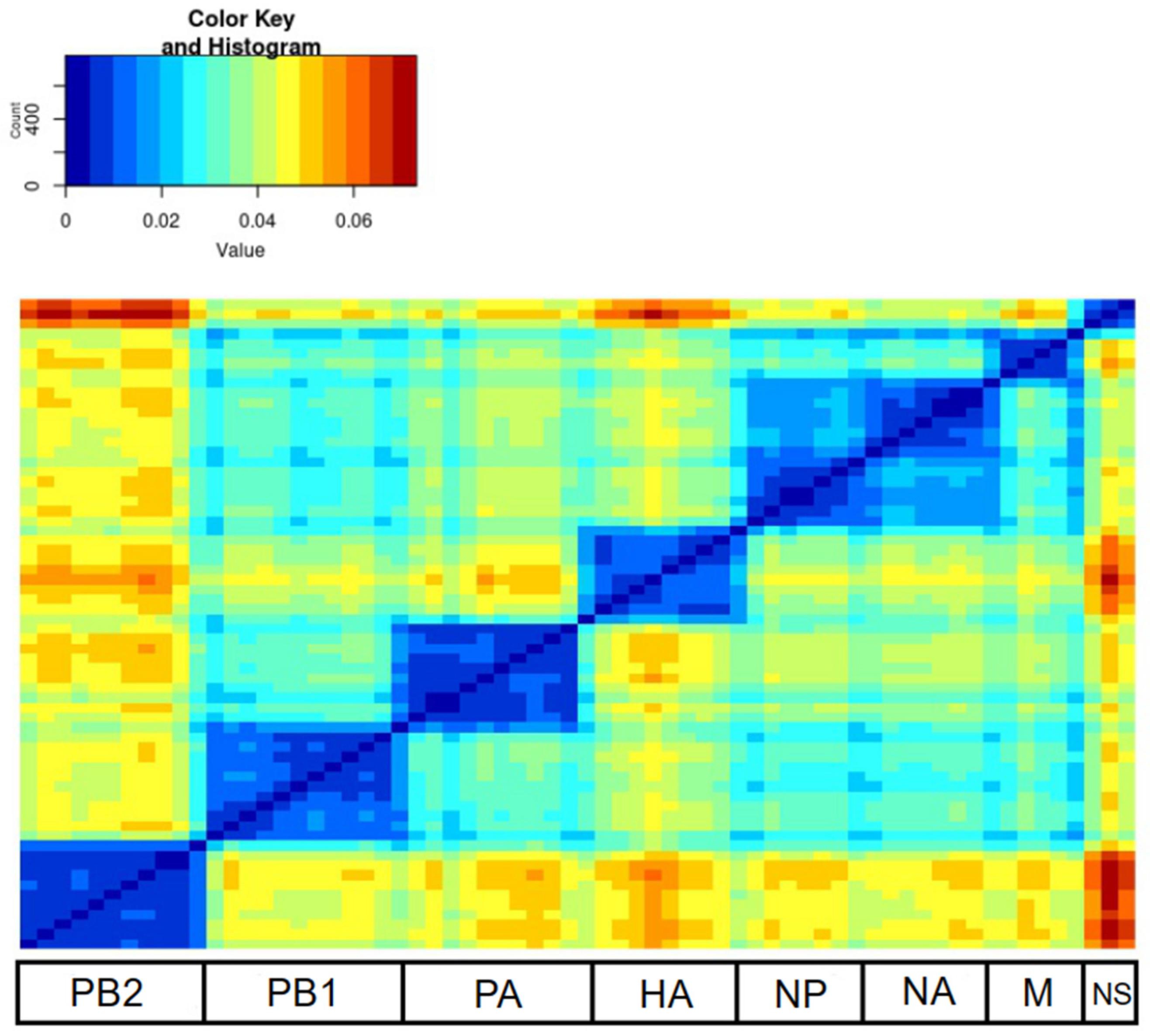
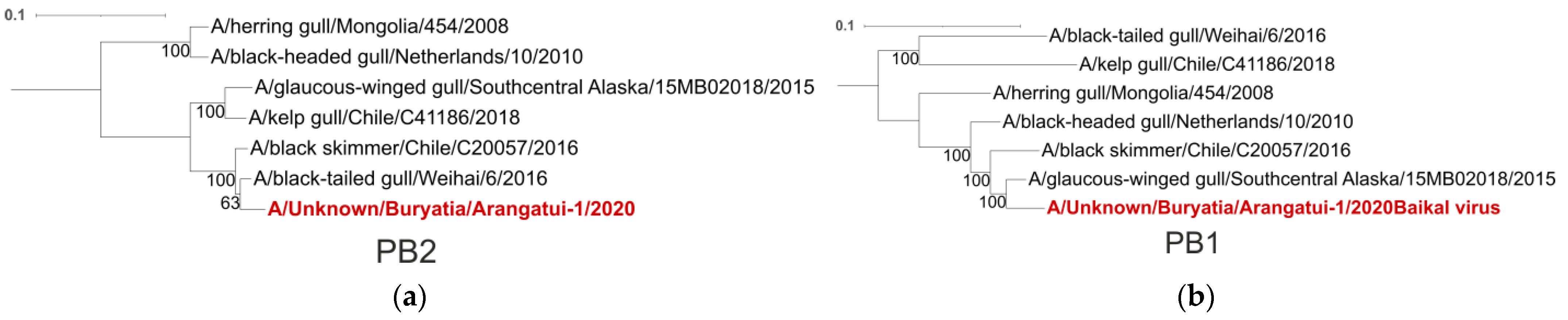
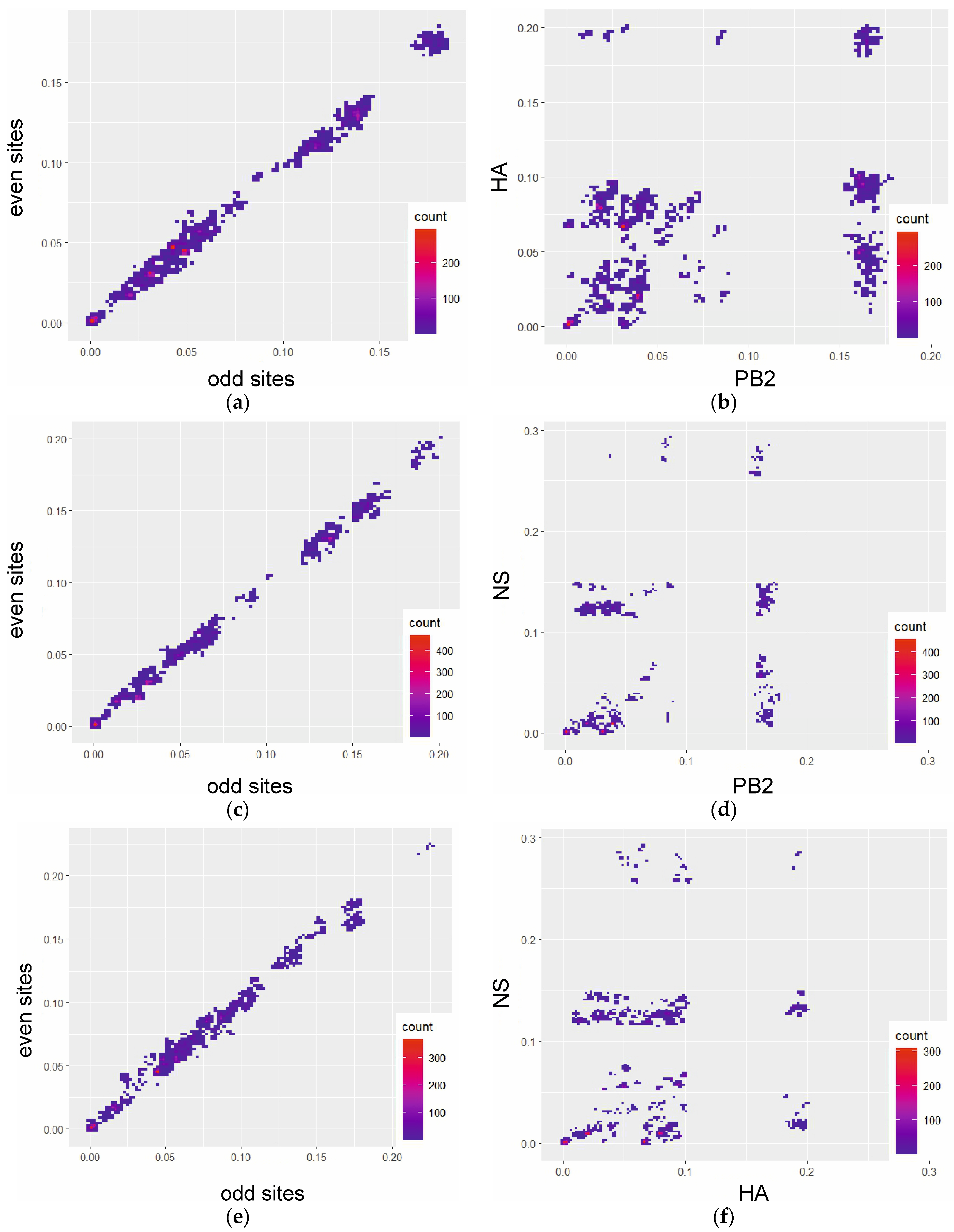
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martini, M.; Gazzaniga, V.; Bragazzi, N.L.; Barberis, I. The Spanish Influenza Pandemic: A Lesson from History 100 Years after 1918. J. Prev. Med. Hyg. 2019, 60, E64–E67. [Google Scholar] [CrossRef] [PubMed]
- Creytens, S.; Pascha, M.N.; Ballegeer, M.; Saelens, X.; de Haan, C.A.M. Influenza Neuraminidase Characteristics and Potential as a Vaccine Target. Front. Immunol. 2021, 12, 786617. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Wilson, I.A. Influenza Hemagglutinin Structures and Antibody Recognition. Cold Spring Harb. Perspect. Med. 2020, 10, a038778. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Prim. 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Fereidouni, S.; Starick, E.; Karamendin, K.; Di Genova, C.; Scott, S.D.; Khan, Y.; Harder, T.; Kydyrmanov, A. Genetic Characterization of a New Candidate Hemagglutinin Subtype of Influenza A Viruses. Emerg. Microbes Infect. 2023, 12, 2225645. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and Ecology of Influenza A Viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-T.; Linster, M.; Mendenhall, I.H.; Su, Y.C.F.; Smith, G.J.D. Avian Influenza Viruses in Humans: Lessons from Past Outbreaks. Br. Med. Bull. 2019, 132, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Blagodatski, A.; Trutneva, K.; Glazova, O.; Mityaeva, O.; Shevkova, L.; Kegeles, E.; Onyanov, N.; Fede, K.; Maznina, A.; Khavina, E.; et al. Avian Influenza in Wild Birds and Poultry: Dissemination Pathways, Monitoring Methods, and Virus Ecology. Pathogens 2021, 10, 630. [Google Scholar] [CrossRef]
- Crisci, E.; Mussá, T.; Fraile, L.; Montoya, M. Review: Influenza Virus in Pigs. Mol. Immunol. 2013, 55, 200–211. [Google Scholar] [CrossRef]
- Campos, A.C.A.; Góes, L.G.B.; Moreira-Soto, A.; de Carvalho, C.; Ambar, G.; Sander, A.-L.; Fischer, C.; Ruckert da Rosa, A.; Cardoso de Oliveira, D.; Kataoka, A.P.G.; et al. Bat Influenza A(HL18NL11) Virus in Fruit Bats, Brazil. Emerg. Infect. Dis. 2019, 25, 333–337. [Google Scholar] [CrossRef]
- Borland, S.; Gracieux, P.; Jones, M.; Mallet, F.; Yugueros-Marcos, J. Influenza A Virus Infection in Cats and Dogs: A Literature Review in the Light of the “One Health” Concept. Front. Public Health 2020, 8, 83. [Google Scholar] [CrossRef]
- Runstadler, J.A.; Puryear, W. A Brief Introduction to Influenza A Virus in Marine Mammals. Methods Mol. Biol. 2020, 2123, 429–450. [Google Scholar]
- Hall, J.S.; TeSlaa, J.L.; Nashold, S.W.; Halpin, R.A.; Stockwell, T.; Wentworth, D.E.; Dugan, V.; Ip, H.S. Evolution of a Reassortant North American Gull Influenza Virus Lineage: Drift, Shift and Stability. Virol. J. 2013, 10, 179. [Google Scholar] [CrossRef]
- Wille, M.; Robertson, G.J.; Whitney, H.; Bishop, M.A.; Runstadler, J.A.; Lang, A.S. Extensive Geographic Mosaicism in Avian Influenza Viruses from Gulls in the Northern Hemisphere. PLoS ONE 2011, 6, e20664. [Google Scholar] [CrossRef]
- Dusek, R.J.; Hallgrimsson, G.T.; Ip, H.S.; Jónsson, J.E.; Sreevatsan, S.; Nashold, S.W.; TeSlaa, J.L.; Enomoto, S.; Halpin, R.A.; Lin, X.; et al. North Atlantic Migratory Bird Flyways Provide Routes for Intercontinental Movement of Avian Influenza Viruses. PLoS ONE 2014, 9, e92075. [Google Scholar] [CrossRef]
- Lindsay, L.L.; Plancarte, M.; Brenn-White, M.; Boyce, W.M. Complete Genome Sequences of the First Reported California H16 Influenza A Viruses. Genome Announc. 2014, 2, e00345-14. [Google Scholar] [CrossRef]
- Lindh, E.; Ek-Kommonen, C.; Isomursu, M.; Alasaari, J.; Vaheri, A.; Vapalahti, O.; Huovilainen, A. Genetic characterization of H13 and h16 influenza a viruses in gulls (Larus spp.) with clinically severe disease and concurrent circovirus infection. J. Wildl. Dis. 2017, 53, 561–571. [Google Scholar] [CrossRef]
- Yu, Z.; He, H.; Cheng, K.; Wu, J.; Gao, Y.; Chen, W.; Yuan, X.; Zhao, Y. Genetic Characterization of an H13N2 Low Pathogenic Avian Influenza Virus Isolated from Gulls in China. Transbound. Emerg. Dis. 2019, 66, 1063–1066. [Google Scholar] [CrossRef]
- Verhagen, J.H.; Poen, M.; Stallknecht, D.E.; van der Vliet, S.; Lexmond, P.; Sreevatsan, S.; Poulson, R.L.; Fouchier, R.A.M.; Lebarbenchon, C. Phylogeography and Antigenic Diversity of Low-Pathogenic Avian Influenza H13 and H16 Viruses. J. Virol. 2020, 94, e00537-20. [Google Scholar] [CrossRef]
- WHO Influenza Virus Detection Protocols. Available online: https://cdn.who.int/media/docs/default-source/influenza/molecular-detention-of-influenza-viruses/protocols_influenza_virus_detection_feb_2021.pdf?sfvrsn=df7d268a_5 (accessed on 7 February 2021).
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A Better Web Interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, A.O.; Volchkov, P.Y.; Deviatkin, A.A. Concatenation of Segmented Viral Genomes for Reassortment Analysis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Vakulenko, Y.; Deviatkin, A.; Drexler, J.F.; Lukashev, A. Modular Evolution of Coronavirus Genomes. Viruses 2021, 13, 1270. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A Computer Program for Analyzing Recombination in, and Removing Signals of Recombination from, Nucleotide Sequence Datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Samson, S.; Lord, É.; Makarenkov, V. SimPlot++: A Python Application for Representing Sequence Similarity and Detecting Recombination. Bioinformatics 2022, 38, 3118–3120. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. Corrigendum to: IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 2461. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2018, 20, 1160–1166. [Google Scholar] [CrossRef]
- Meng, B.; Wang, Q.; Leng, H.; Ren, C.; Feng, C.; Guo, W.; Feng, Y.; Zhang, Y. Evolutionary Events Promoted Polymerase Activity of H13N8 Avian Influenza Virus. Viruses 2024, 16, 329. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.J.; Bishop, M.A.; Trovão, N.S.; Ineson, K.M.; Schaefer, A.L.; Puryear, W.B.; Zhou, K.; Foss, A.D.; Clark, D.E.; MacKenzie, K.G.; et al. Ecological Divergence of Wild Birds Drives Avian Influenza Spillover and Global Spread. PLOS Pathog. 2022, 18, e1010062. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhao, M.; Yu, Z.; Li, Y.; Zhang, X.; Feng, N.; Wang, T.; Wang, H.; He, H.; Zhao, Y.; et al. Cross-Species Infection Potential of Avian Influenza H13 Viruses Isolated from Wild Aquatic Birds to Poultry and Mammals. Emerg. Microbes Infect. 2023, 12, e2184177. [Google Scholar] [CrossRef] [PubMed]
- De Marco, M.A.; Delogu, M.; Facchini, M.; Di Trani, L.; Boni, A.; Cotti, C.; Graziosi, G.; Venturini, D.; Regazzi, D.; Ravaioli, V.; et al. Serologic Evidence of Occupational Exposure to Avian Influenza Viruses at the Wildfowl/Poultry/Human Interface. Microorganisms 2021, 9, 2153. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I.; Si, Y.J.; Kwon, H.I.; Kim, E.H.; Park, S.J.; Robles, N.J.; Nguyen, H.D.; Yu, M.A.; Yu, K.M.; Lee, Y.J.; et al. Pathogenicity and Genetic Characterisation of a Novel Reassortant, Highly Pathogenic Avian Influenza (HPAI) H5N6 Virus Isolated in Korea, 2017. Eurosurveillance 2018, 23, 1–8. [Google Scholar] [CrossRef]
- Huang, P.; Sun, L.; Li, J.; Wu, Q.; Rezaei, N.; Jiang, S.; Pan, C. Potential Cross-Species Transmission of Highly Pathogenic Avian Influenza H5 Subtype (HPAI H5) Viruses to Humans Calls for the Development of H5-Specific and Universal Influenza Vaccines. Cell Discov. 2023, 9, 58. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
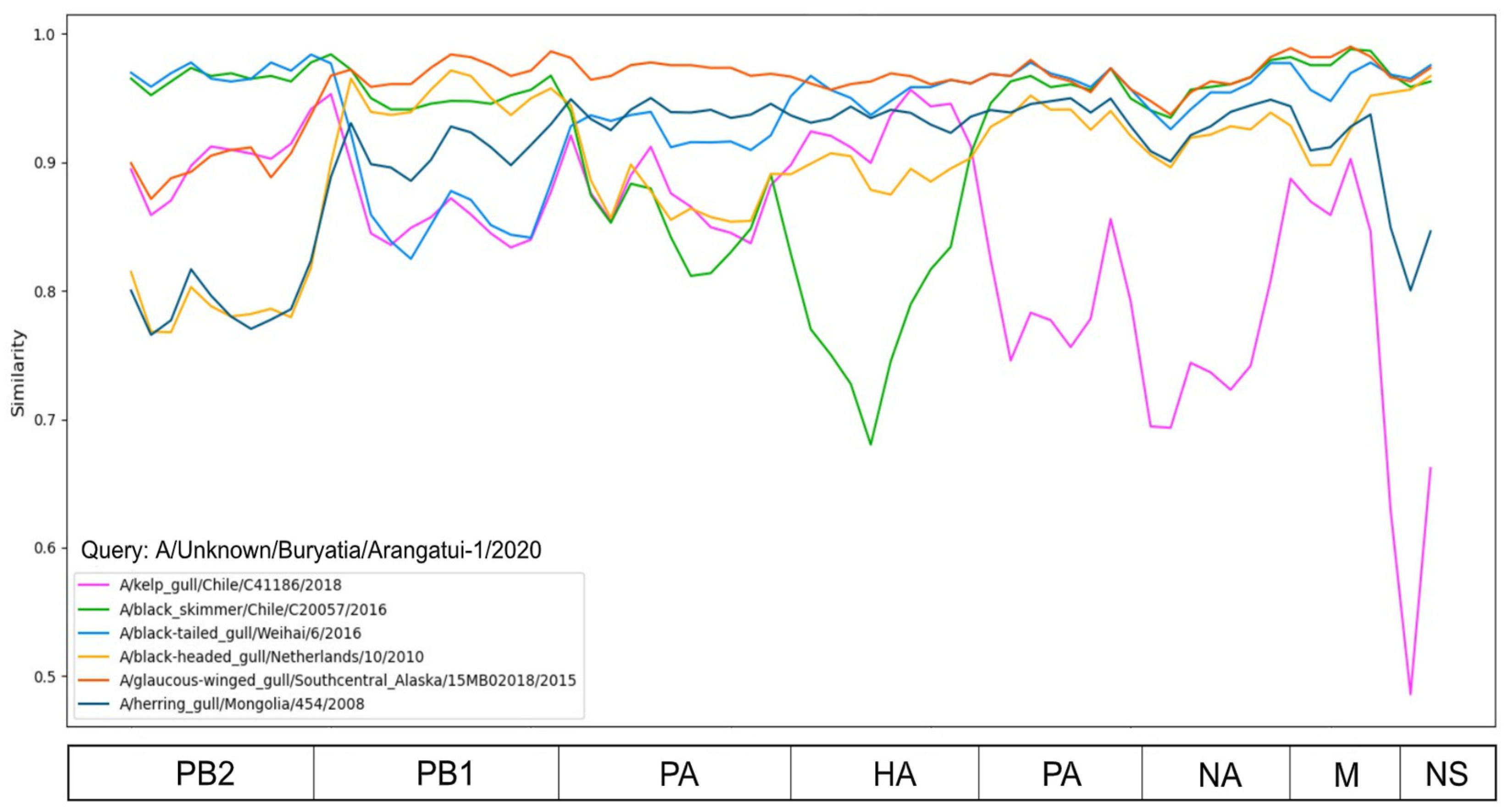
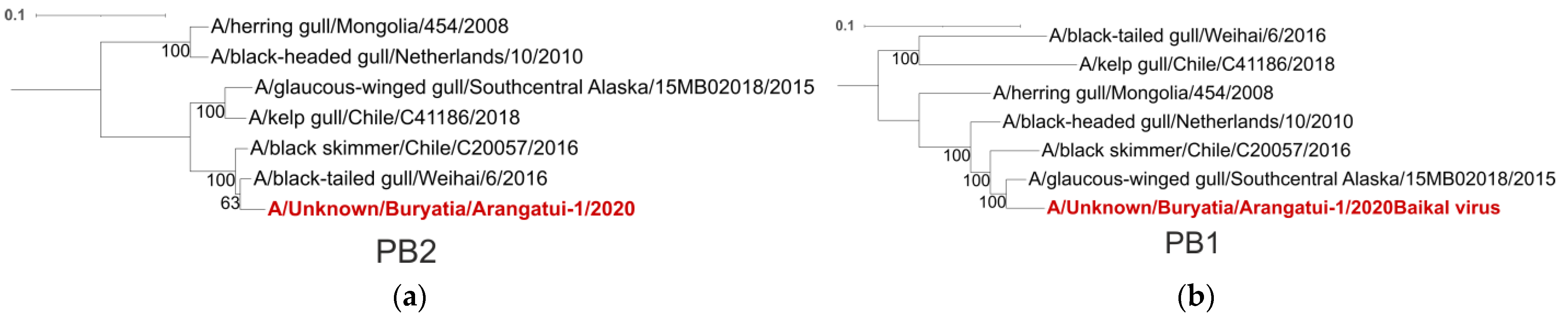
| Segment | Closest Viruses | Place of Collection |
|---|---|---|
| PB2 | A/Black-tailed gull/Weihai/6/2016 | China |
| A/Black skimmer/Chile/C20057/2016 | Chile | |
| PB1 | A/Glaucus-winged gull/Southcentral Alaska/15MB02018/2015 | USA |
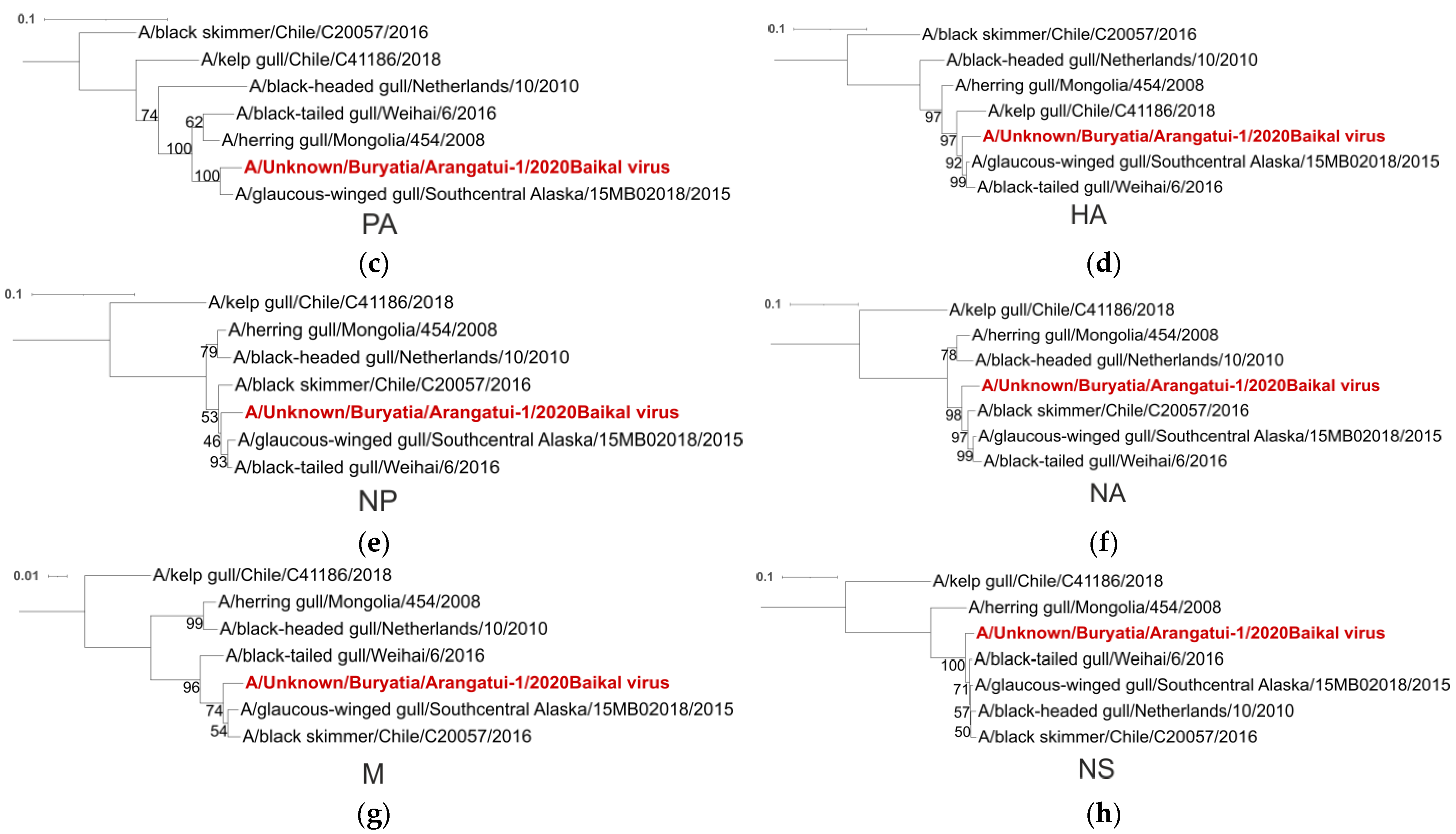
| PA | A/Glaucus-winged gull/Southcentral Alaska/15MB02018/2015 | USA |
| HA | A/Glaucus-winged gull/Southcentral Alaska/15MB02018/2015 | USA |
| A/Black-tailed gull/Weihai/6/2016 | China | |
| NP | A/Glaucus-winged gull/Southcentral Alaska/15MB02018/2015 | USA |
| A/Black-tailed gull/Weihai/6/2016 | China | |
| A/Black skimmer/Chile/C20057/2016 | Chile | |
| NA | A/Glaucus-winged gull/Southcentral Alaska/15MB02018/2015 | USA |
| A/Black-tailed gull/Weihai/6/2016 | China | |
| A/Black skimmer/Chile/C20057/2016 | Chile | |
| M | A/Glaucus-winged gull/Southcentral Alaska/15MB02018/2015 | USA |
| A/Black skimmer/Chile/C20057/2016 | Chile | |
| NS | A/Glaucus-winged gull/Southcentral Alaska/15MB02018/2015 | USA |
| A/Black-tailed gull/Weihai/6/2016 | China | |
| A/Black skimmer/Chile/C20057/2016 | Chile | |
| A/Black-headed gull/Netherlands/10/2010 | Netherlands |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feoktistova, S.; Sayganova, M.; Trutneva, K.; Glazova, O.; Blagodatski, A.S.; Shevkova, L.; Navoikova, A.; Anisimov, Y.; Albert, E.; Mityaeva, O.; et al. Abundant Intra-Subtype Reassortment Revealed in H13N8 Influenza Viruses. Viruses 2024, 16, 568. https://doi.org/10.3390/v16040568
Feoktistova S, Sayganova M, Trutneva K, Glazova O, Blagodatski AS, Shevkova L, Navoikova A, Anisimov Y, Albert E, Mityaeva O, et al. Abundant Intra-Subtype Reassortment Revealed in H13N8 Influenza Viruses. Viruses. 2024; 16(4):568. https://doi.org/10.3390/v16040568
Chicago/Turabian StyleFeoktistova, Sofia, Marya Sayganova, Kseniya Trutneva, Olga Glazova, Artem S. Blagodatski, Liudmila Shevkova, Anna Navoikova, Yuriy Anisimov, Eugene Albert, Olga Mityaeva, and et al. 2024. "Abundant Intra-Subtype Reassortment Revealed in H13N8 Influenza Viruses" Viruses 16, no. 4: 568. https://doi.org/10.3390/v16040568