
Improving Pharmacokinetics of Peptides Using Phage Display

Abstract
:1. Introduction
2. Filamentous Bacteriophages
3. Principles of Phage Display
3.1. Phage Display Systems
3.2. Bacteriophage Vectors and Phagemids
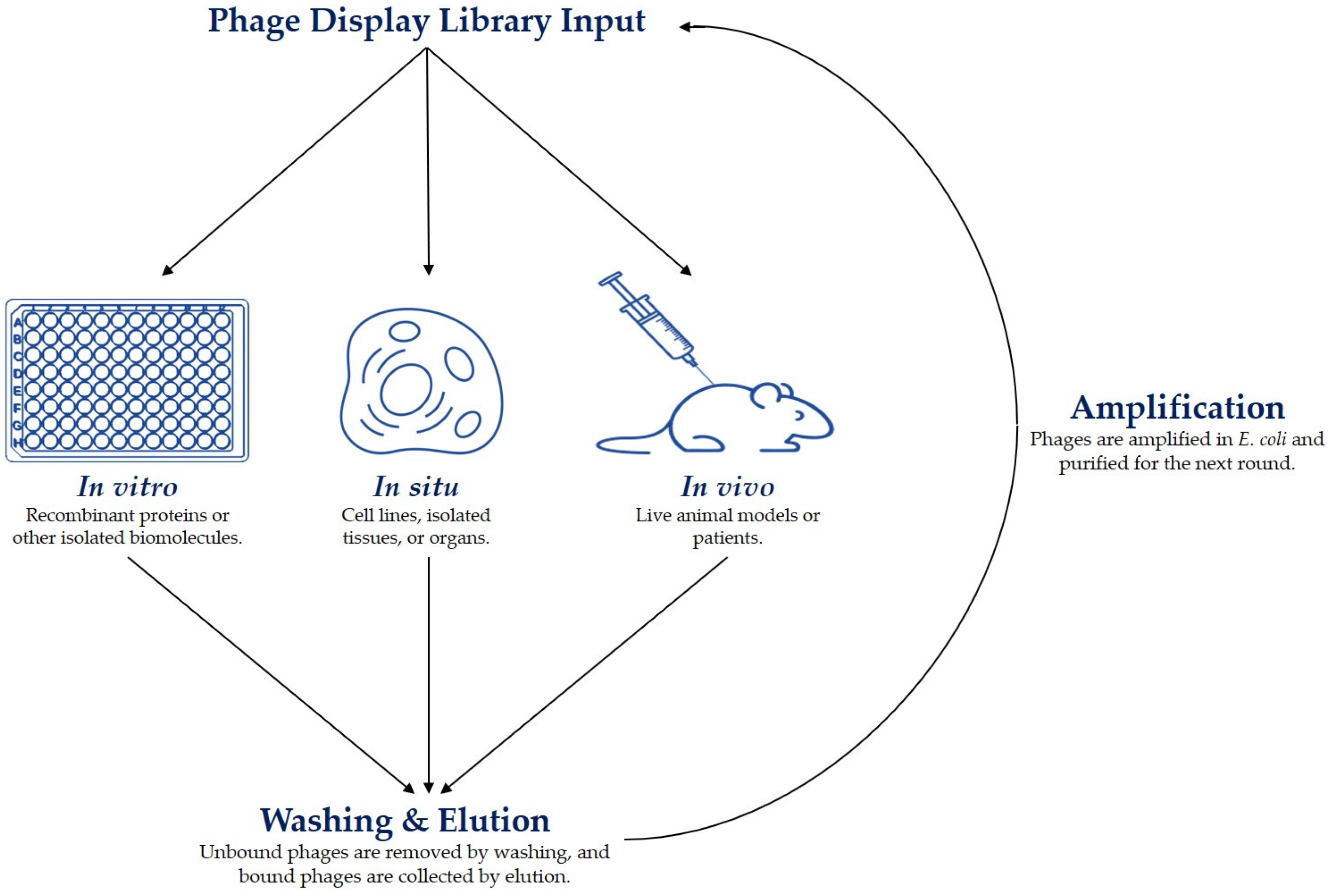
3.3. Affinity Selection (Biopanning)
3.4. Peptide Phage Display
3.4.1. In Vitro Selected Peptides
3.4.2. In Situ Selected Peptides
3.4.3. In Vivo Selected Peptides
3.4.4. Peptide-Conjugated Nanoparticles
4. Pharmacokinetic Principles
4.1. Pharmacokinetics of Peptides
4.1.1. Routes of Administration
4.1.2. Distribution and Absorption
4.1.3. Clearance
4.1.4. Drug-Specific Issues
4.2. Pharmacokinetics of Phage and Nanoparticles
4.2.1. Pharmacokinetic Profile of Filamentous Phage
4.2.2. Distribution and Absorption
4.2.3. Clearance
4.2.4. Effect of Size, Shape, and Charge upon Phage Pharmacokinetics
4.2.5. Pharmacodynamics of Phage Display Selected Peptides
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar] [CrossRef]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef]
- França, R.K.A.; Studart, I.C.; Bezerra, M.R.L.; Pontes, L.Q.; Barbosa, A.M.A.; Brigido, M.M.; Furtado, G.P.; Maranhão, A.Q. Progress on phage display technology: Tailoring antibodies for cancer immunotherapy. Viruses 2023, 15, 1903. [Google Scholar] [CrossRef]
- Islam, M.S.; Fan, J.; Pan, F. The power of phages: Revolutionizing cancer treatment. Front. Oncol. 2023, 13, 1290296. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Morcinek-Orłowska, J.; Gaffke, L.; Jaroszewicz, W.; Skowron, P.M.; Węgrzyn, G. Applications of the phage display technology in molecular biology, biotechnology and medicine. Crit. Rev. Microbiol. 2023, 1–41. [Google Scholar] [CrossRef]
- Song, B.P.C.; Ch’ng, A.C.W.; Lim, T.S. Review of phage display: A jack-of-all-trades and master of most biomolecule display. Int. J. Biol. Macromol. 2024, 256, 128455. [Google Scholar] [CrossRef]
- Arab, A.; Nicastro, J.; Slavcev, R.; Razazan, A.; Barati, N.; Nikpoor, A.R.; Brojeni, A.A.M.; Mosaffa, F.; Badiee, A.; Jaafari, M.R.; et al. Lambda phage nanoparticles displaying her2-derived e75 peptide induce effective e75-cd8(+) t response. Immunol. Res. 2018, 66, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Barderas, R.; Benito-Pena, E. The 2018 nobel prize in chemistry: Phage display of peptides and antibodies. Anal. Bioanal. Chem. 2019, 411, 2475–2479. [Google Scholar] [CrossRef]
- Voulgaridou, G.P.; Theologidis, V.; Xanthis, V.; Papagiannaki, E.; Tsochantaridis, I.; Fadouloglou, V.E.; Pappa, A. Identification of a peptide ligand for human aldh3a1 through peptide phage display: Prediction and characterization of protein interaction sites and inhibition of aldh3a1 enzymatic activity. Front. Mol. Biosci. 2023, 10, 1161111. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Wu, J.; Wang, Y.; Wu, Y.; Sun, X.; Wang, X.; Shen, J.; Xie, L.; Zhang, Y.; et al. Linear peptide-based pet tracers for imaging pd-l1 in tumors. Mol. Pharm. 2023, 20, 4256–4267. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Principles of affinity selection. Cold Spring Harb. Protoc. 2023. [Google Scholar] [CrossRef]
- Deutscher, S.L. Phage display in molecular imaging and diagnosis of cancer. Chem. Rev. 2010, 110, 3196–3211. [Google Scholar] [CrossRef]
- Arap, W.; Kolonin, M.G.; Trepel, M.; Lahdenranta, J.; Cardo-Vila, M.; Giordano, R.J.; Mintz, P.J.; Ardelt, P.U.; Yao, V.J.; Vidal, C.I.; et al. Steps toward mapping the human vasculature by phage display. Nat. Med. 2002, 8, 121–127. [Google Scholar] [CrossRef]
- Yao, V.J.; Ozawa, M.G.; Trepel, M.; Arap, W.; McDonald, D.M.; Pasqualini, R. Targeting pancreatic islets with phage display assisted by laser pressure catapult microdissection. Am. J. Pathol. 2005, 166, 625–636. [Google Scholar] [CrossRef]
- Yao, J.F.; Yang, H.; Zhao, Y.Z.; Xue, M. Metabolism of peptide drugs and strategies to improve their metabolic stability. Curr. Drug Metab. 2018, 19, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Eden, H.S.; Chen, X. Peptides in cancer nanomedicine: Drug carriers, targeting ligands and protease substrates. J. Control Release 2012, 159, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.C. Peptidic tumor targeting agents: The road from phage display peptide selections to clinical applications. Curr. Pharm. Des. 2010, 16, 1040–1054. [Google Scholar] [CrossRef]
- Kolodziej, A.F.; Zhang, Z.; Overoye-Chan, K.; Jacques, V.; Caravan, P. Peptide optimization and conjugation strategies in the development of molecularly targeted magnetic resonance imaging contrast agents. Methods Mol. Biol. 2014, 1088, 185–211. [Google Scholar] [CrossRef]
- Chen, S.; Gfeller, D.; Buth, S.A.; Michielin, O.; Leiman, P.G.; Heinis, C. Improving binding affinity and stability of peptide ligands by substituting glycines with d-amino acids. Chembiochem 2013, 14, 1316–1322. [Google Scholar] [CrossRef]
- Ngambenjawong, C.; Pineda, J.M.; Pun, S.H. Engineering an affinity-enhanced peptide through optimization of cyclization chemistry. Bioconjug. Chem. 2016, 27, 2854–2862. [Google Scholar] [CrossRef]
- Soendergaard, M.; Newton-Northup, J.R.; Deutscher, S.L. In vivo phage display selection of an ovarian cancer targeting peptide for spect/ct imaging. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 561–570. [Google Scholar]
- Hou, L.D.; Zhu, D.X.; Liang, Y.; Tian, X.H.; Li, L.; Wang, P.; Zhu, L.M.; Weng, X.L.; Wang, Y.Y.; Li, Y.; et al. Identification of a specific peptide binding to colon cancer cells from a phage-displayed peptide library. Br. J. Cancer 2018, 118, 79–87. [Google Scholar] [CrossRef]
- Zhang, D.; Huang, J.; Li, W.M.; Zhang, Z.Y.; Zhu, M.; Feng, Y.; Zhao, Y.; Li, Y.R.; Lu, S.Y.; He, S.X. Screening and identification of a cd44v6 specific peptide using improved phage display for gastric cancer targeting. Ann. Transl. Med. 2020, 8, 1442. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Gao, X.J.; Xiao, L.; He, H.M.; Cheng, S.N.; Zhang, C.X.; Hou, Y.F.; Song, F.Y.; Su, X.R.; Gao, Q.; et al. Screening and identification of a specific peptide binding to breast cancer cells from a phage-displayed peptide library. Biotechnol. Lett. 2021, 43, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Zhu, R.J.; Hu, W.; Zhang, W.; Zhu, H.B.; Gong, M.; Gao, L.L.; Cao, T.; Tang, T.; Yu, B.; et al. Phage display screening identifies a prostate specific antigen (psa)(-/lo) prostate cancer cell specific peptide to retard castration resistance of prostate cancer. Transl. Oncol. 2021, 14, 101020. [Google Scholar] [CrossRef]
- Sioud, M.; Zhang, Q.D. Precision killing of m2 macrophages with phage-displayed peptide-photosensitizer conjugates. Cancers 2023, 15, 2009. [Google Scholar] [CrossRef] [PubMed]
- Andre, A.S.; Moutinho, I.; Dias, J.N.R.; Aires-da-Silva, F. In vivo phage display: A promising selection strategy for the improvement of antibody targeting and drug delivery properties. Front. Microbiol. 2022, 13, 962124. [Google Scholar] [CrossRef] [PubMed]
- Pleiko, K.; Põšnograjeva, K.; Haugas, M.; Paiste, P.; Tobi, A.; Kurm, K.; Riekstina, U.; Teesalu, T. In vivo phage display: Identification of organ-specific peptides using deep sequencing and differential profiling across tissues. Nucleic Acids Res. 2021, 49, e38. [Google Scholar] [CrossRef]
- Aguiar, S.I.; Dias, J.N.R.; Andre, A.S.; Silva, M.L.; Martins, D.; Carrapico, B.; Castanho, M.; Carrico, J.; Cavaco, M.; Gaspar, M.M.; et al. Highly specific blood-brain barrier transmigrating single-domain antibodies selected by an in vivo phage display screening. Pharmaceutics 2021, 13, 1598. [Google Scholar] [CrossRef]
- Qin, Y.S.; Cheng, S.Y.; Li, Y.S.; Zou, S.J.; Chen, M.L.; Zhu, D.L.; Gao, S.; Wu, H.; Zhu, L.; Zhu, X.H. The development of a glypican-3-specific binding peptide usingin vivoandin vitrotwo-step phage display screening for the pet imaging of hepatocellular carcinoma. Biomater. Sci. 2020, 8, 5656–5665. [Google Scholar] [CrossRef]
- Jaroszewicz, W.; Morcinek-Orlowska, J.; Pierzynowska, K.; Gaffke, L.; Wegrzyn, G. Phage display and other peptide display technologies. FEMS Microbiol. Rev. 2022, 46, fuab052. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Liu, M.; Xie, S.B. Harnessing phage display for the discovery of peptide-based drugs and monoclonal antibodies. Curr. Med. Chem. 2021, 28, 8267–8274. [Google Scholar] [CrossRef] [PubMed]
- Zambrano-Mila, M.S.; Blacio, K.E.S.; Vispo, N.S. Peptide phage display: Molecular principles and biomedical applications. Ther. Innov. Regul. Sci. 2020, 54, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Rakonjac, J.; Russel, M.; Khanum, S.; Brooke, S.J.; Rajic, M. Filamentous phage: Structure and biology. Adv. Exp. Med. Biol. 2017, 1053, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Barbas, C.F.; Burton, D.R.; Scott, J.K.; Fraser, S.; Silverman, G.J. Filamentous phage biology. In Phage Display: A Laboratory Manual; Cold Spring Harber Laboratory Press: Woodbury, NY, USA, 2001; pp. 1.1–1.37. [Google Scholar]
- Hess, K.L.; Jewell, C.M. Phage display as a tool for vaccine and immunotherapy development. Bioeng. Transl. Med. 2020, 5, e10142. [Google Scholar] [CrossRef] [PubMed]
- Marintcheva, B.; ScienceDirect. Harnessing the Power of Viruses; Academic Press: London, UK, 2017. [Google Scholar]
- Loh, B.; Kuhn, A.; Leptihn, S. The fascinating biology behind phage display: Filamentous phage assembly. Mol. Microbiol. 2019, 111, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Phage display libraries: From binders to targeted drug delivery and human therapeutics. Mol. Biotechnol. 2019, 61, 286–303. [Google Scholar] [CrossRef]
- Omidfar, K.; Daneshpour, M. Advances in phage display technology for drug discovery. Expert Opin. Drug Discov. 2015, 10, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gacio, A.; Uguen, M.; Fastrez, J. Phage display as a tool for the directed evolution of enzymes. Trends Biotechnol. 2003, 21, 408–414. [Google Scholar] [CrossRef]
- Hamzeh-Mivehroud, M.; Alizadeh, A.A.; Morris, M.B.; Church, W.B.; Dastmalchi, S. Phage display as a technology delivering on the promise of peptide drug discovery. Drug Discov. Today 2013, 18, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Scott, J.K. [15] libraries of peptides and proteins displayed on filamentous phage. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1993; Volume 217, pp. 228–257. [Google Scholar]
- Arap, M.A. Phage display technology: Applications and innovations. Genet. Mol. Biol. 2005, 28, 1–9. [Google Scholar] [CrossRef]
- Enshell-Seijffers, D.; Smelyanski, L.; Gershoni, J.M. The rational design of a ‘type 88’ genetically stable peptide display vector in the filamentous bacteriophage fd. Nucleic Acids Res. 2001, 29, E50. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar] [CrossRef]
- Malik, P.; Terry, T.D.; Bellintani, F.; Perham, R.N. Factors limiting display of foreign peptides on the major coat protein of filamentous bacteriophage capsids and a potential role for leader peptidase. FEBS Lett. 1998, 436, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Jończyk, E.; Kłak, M.; Międzybrodzki, R.; Górski, A. The influence of external factors on bacteriophages—Review. Folia Microbiol. 2011, 56, 191–200. [Google Scholar] [CrossRef]
- Jespers, L.S.; Messens, J.H.; De Keyser, A.; Eeckhout, D.; Van den Brande, I.; Gansemans, Y.G.; Lauwereys, M.J.; Vlasuk, G.P.; Stanssens, P.E. Surface expression and ligand-based selection of cdnas fused to filamentous phage gene vi. Biotechnology 1995, 13, 378–382. [Google Scholar] [CrossRef]
- Gao, X.; Huang, Y.; Zhu, S. Construction of murine phage antibody library and selection of ricin-specific single-chain antibodies. IUBMB Life 1999, 48, 513–517. [Google Scholar]
- Løset, G.Å.; Bogen, B.; Sandlie, I. Expanding the versatility of phage display i: Efficient display of peptide-tags on protein vii of the filamentous phage. PLoS ONE 2011, 6, e14702. [Google Scholar] [CrossRef]
- Løset, G.; Roos, N.; Bogen, B.; Sandlie, I. Expanding the versatility of phage display ii: Improved affinity selection of folded domains on protein vii and ix of the filamentous phage. PLoS ONE 2011, 6, e17433. [Google Scholar] [CrossRef]
- Smith, G.P. Preface. Gene 1993, 128, 1–2. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Seong, B.L. Solubility, stability, and avidity of recombinant antibody fragments expressed in microorganisms. Front. Microbiol. 2020, 11, 1927. [Google Scholar] [CrossRef]
- Qi, H.; Lu, H.; Qiu, H.-J.; Petrenko, V.; Liu, A. Phagemid vectors for phage display: Properties, characteristics and construction. J. Mol. Biol. 2012, 417, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Rader, C. The pcomb3 phagemid family of phage display vectors. Cold Spring Harb. Protoc. 2023. [Google Scholar] [CrossRef]
- Ehrlich, G.K.; Berthold, W.; Bailon, P. Phage display technology. Affinity selection by biopanning. Methods Mol. Biol. 2000, 147, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, H.; Umetsu, M.; Tatsuya, H.; Hattori, T.; Kumagai, I. Identification of indium tin oxide nartoparticle-biliding peptides via phage display and biopanning under various buffer conditions. Protein Pept. Lett. 2020, 27, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.A.; Bachtiger, F.; Kong, X.D.; Abriata, L.A.; Sosso, G.C.; Gibson, M.I.; Klok, H.A. A minimalistic cyclic ice-binding peptide from phage display. Nat. Commun. 2021, 12, 2675. [Google Scholar] [CrossRef]
- Landon, L.A.; Peletskaya, E.N.; Glinsky, V.V.; Karasseva, N.; Quinn, T.P.; Deutscher, S.L. Combinatorial evolution of high-affinity peptides that bind to the thomsen-friedenreich carcinoma antigen. J. Protein Chem. 2003, 22, 193–204. [Google Scholar] [CrossRef]
- Li, J.; Feng, L.; Jiang, X. In vivo phage display screen for peptide sequences that cross the blood-cerebrospinal-fluid barrier. Amino Acids 2015, 47, 401–405. [Google Scholar] [CrossRef]
- Li, X.; Ma, Z.; Wang, H.R.; Ren, L.; Zhang, D.W.; Liang, W.G.; Zhang, G.J.; Zhang, J.R.; Yu, D.H.; Fang, X.X. Screening, identification, and characterization of an affinity peptide specific to mt1-mmp and its application in tumor imaging. Bioconjug. Chem. 2019, 30, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Northup, J.R.N.; Deutscher, S.L. Cytotoxic tumor-targeting peptides from in vivo phage display. Comb. Chem. High. Throughput Screen. 2016, 19, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Shang, W.T.; Guo, P.Y.; He, K.S.; Wang, H.Z.; Han, Z.Y.; Jiang, H.M.; Tian, J.; Wang, K.; Xu, W.H. Phage display-derived peptide-based dual-modality imaging probe for bladder cancer diagnosis and resection postinstillation: A preclinical study. Mol. Cancer Ther. 2018, 17, 2100–2111. [Google Scholar] [CrossRef] [PubMed]
- Loset, G.A.; Kristinsson, S.G.; Sandlie, I. Reliable titration of filamentous bacteriophages independent of pIII fusion moiety and genome size by using trypsin to restore wild-type piii phenotype. Biotechniques 2008, 44, 551–552, 554. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.D.; Smith, G.P. The case for trypsin release of affinity-selected phages. BioTechniques 2010, 49, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Plessers, S.; Van Deuren, V.; Lavigne, R.; Robben, J. High-throughput sequencing of phage display libraries reveals parasitic enrichment of indel mutants caused by amplification bias. Int. J. Mol. Sci. 2021, 22, 5513. [Google Scholar] [CrossRef] [PubMed]
- Sloth, A.B.; Bakhshinejad, B.; Jensen, M.; Stavnsbjerg, C.; Liisberg, M.B.; Rossing, M.; Kjaer, A. Analysis of compositional bias in a commercial phage display peptide library by next-generation sequencing. Viruses 2022, 14, 2402. [Google Scholar] [CrossRef] [PubMed]
- Van Deuren, V.; Plessers, S.; Lavigne, R.; Robben, J. Application of Deep Sequencing in Phage Display; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2024; Volume 2738, pp. 333–345. [Google Scholar] [CrossRef]
- Spiliotopoulos, A.; Maurer, S.K.; Tsoumpeli, M.T.; Bonfante, J.A.F.; Owen, J.P.; Gough, K.C.; Dreveny, I. Next-generation phage display to identify peptide ligands of deubiquitinases. Methods Mol. Biol. 2023, 2591, 189–218. [Google Scholar] [CrossRef]
- Vaisman-Mentesh, A.; Wine, Y. Monitoring phage biopanning by next-generation sequencing. Methods Mol. Biol. 2018, 1701, 463–473. [Google Scholar] [CrossRef]
- Bratkovic, T. Progress in phage display: Evolution of the technique and its application. Cell Mol. Life Sci. 2010, 67, 749–767. [Google Scholar] [CrossRef]
- Szardenings, M. Phage display of random peptide libraries: Applications, limits, and potential. J. Recept. Signal Transduct. Res. 2003, 23, 307–349. [Google Scholar] [CrossRef] [PubMed]
- Newton-Northup, J.; Duetscher, S.L. Contending with target unrelated peptides from phage display. J. Mol. Imaging Dynam 2012, 2, 1–3. [Google Scholar] [CrossRef]
- Newton-Northup, J.; Deutscher, S.L. Characterization of in vivo selected bacteriophage for the development of novel tumor-targeting agents with specific pharmacokinetics and imaging applications. Methods Mol. Biol. 2017, 1572, 445–465. [Google Scholar] [CrossRef] [PubMed]
- Newton-Northup, J.R.; Dickerson, M.T.; Kumar, S.R.; Smith, G.P.; Quinn, T.P.; Deutscher, S.L. In vivo bacteriophage peptide display to tailor pharmacokinetics of biological nanoparticles. Mol. Imaging Biol. 2014, 16, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Newton-Northup, J.R.; Figueroa, S.D.; Deutscher, S.L. Streamlined in vivo selection and screening of human prostate carcinoma avid phage particles for development of peptide based in vivo tumor imaging agents. Comb. Chem. High. Throughput Screen. 2011, 14, 9–21. [Google Scholar] [CrossRef]
- Trepel, M.; Arap, W.; Pasqualini, R. In vivo phage display and vascular heterogeneity: Implications for targeted medicine. Curr. Opin. Chem. Biol. 2002, 6, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, R.; Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.R.; Kelly, K.A.; Mahmood, U.; Weissleder, R.; Deutscher, S.L. In vivo selection of phage for the optical imaging of pc-3 human prostate carcinoma in mice. Neoplasia 2006, 8, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Deutscher, S.L. Phage peptide display. Handb. Exp. Pharmacol. 2008, 185 Pt 2, 145–163. [Google Scholar] [CrossRef]
- Jin, P.; Sha, R.; Zhang, Y.; Liu, L.; Bian, Y.; Qian, J.; Qian, J.; Lin, J.; Ishimwe, N.; Hu, Y.; et al. Blood circulation-prolonging peptides for engineered nanoparticles identified via phage display. Nano Lett. 2019, 19, 1467–1478. [Google Scholar] [CrossRef]
- Díaz-Perlas, C.; Ricken, B.; Farrera-Soler, L.; Guschin, D.; Pojer, F.; Lau, K.; Gerhold, C.B.; Heinis, C. High-affinity peptides developed against calprotectin and their application as synthetic ligands in diagnostic assays. Nat. Commun. 2023, 14, 2774. [Google Scholar] [CrossRef]
- Zou, J.; Glinsky, V.V.; Landon, L.A.; Matthews, L.; Deutscher, S.L. Peptides specific to the galectin-3 carbohydrate recognition domain inhibit metastasis-associated cancer cell adhesion. Carcinogenesis 2005, 26, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Newton-Northup, J.R.; Dickerson, M.T.; Ma, L.; Besch-Williford, C.L.; Deutscher, S.L. Inhibition of metastatic tumor formation in vivo by a bacteriophage display-derived galectin-3 targeting peptide. Clin. Exp. Metastasis 2013, 30, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, Z.; He, S.; Fan, T.; Jin, Y.; Zhu, X.; Chen, C.; Zhang, Z.R.; Huang, Y. Treatment of prostate carcinoma with (galectin-3)-targeted hpma copolymer-(g3-c12)-5-fluorouracil conjugates. Biomaterials 2012, 33, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, L.; Zhou, Z.; Yang, Q.; Liu, C.; Huang, Y. Targeting prostate carcinoma by g3-c12 peptide conjugated n-(2-hydroxypropyl)methacrylamide copolymers. Mol. Pharm. 2014, 11, 3251–3260. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, L.; Yang, Q.; Shan, W.; Zhang, Z.; Huang, Y. G3-c12 peptide reverses galectin-3 from foe to friend for active targeting cancer treatment. Mol. Pharm. 2015, 12, 4124–4136. [Google Scholar] [CrossRef]
- Kopansky, E.; Shamay, Y.; David, A. Peptide-directed hpma copolymer-doxorubicin conjugates as targeted therapeutics for colorectal cancer. J. Drug Target. 2011, 19, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sahdev, P.; Perumal, O.; Tummala, H. Identification of a novel skin penetration enhancement peptide by phage display peptide library screening. Mol. Pharm. 2012, 9, 1320–1330. [Google Scholar] [CrossRef]
- Kumar, S.R.; Deutscher, S.L. 111in-labeled galectin-3-targeting peptide as a spect agent for imaging breast tumors. J. Nucl. Med. 2008, 49, 796–803. [Google Scholar] [CrossRef]
- Geng, Q.; Sun, X.; Gong, T.; Zhang, Z.R. Peptide-drug conjugate linked via a disulfide bond for kidney targeted drug delivery. Bioconjug. Chem. 2012, 23, 1200–1210. [Google Scholar] [CrossRef]
- Whitney, M.; Crisp, J.L.; Olson, E.S.; Aguilera, T.A.; Gross, L.A.; Ellies, L.G.; Tsien, R.Y. Parallel in vivo and in vitro selection using phage display identifies protease-dependent tumor-targeting peptides. J. Biol. Chem. 2010, 285, 22532–22541. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Pscherer, S.; Gamerdinger, U.; Teigler-Schlegel, A.; Rutz, N.; Blau, W.; Rummel, M.; Gattenlohner, S.; Tur, M.K. Parallel evaluation of cell-based phage display panning strategies: Optimized selection and depletion steps result in aml blast-binding consensus antibodies. Mol. Med. Rep. 2021, 24, 767. [Google Scholar] [CrossRef] [PubMed]
- Asar, M.C.; Franco, A.; Soendergaard, M. Phage display selection, identification, and characterization of novel pancreatic cancer targeting peptides. Biomolecules 2020, 10, 714. [Google Scholar] [CrossRef] [PubMed]
- Anjorin, F.F. Pancreatic cancer specificity of phage display-selected peptide mca1. In Proceedings of the ACS Meeting Spring 2023, Washington, DC, USA, 26–30 March 2023. [Google Scholar]
- Soendergaard, M.; Newton-Northup, J.R.; Deutscher, S.L. In vitro high throughput phage display selection of ovarian cancer avid phage clones for near-infrared optical imaging. Comb. Chem. High. Throughput Screen. 2014, 17, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Dickerson, M.T.; Owen, N.K.; Landon, L.A.; Deutscher, S.L. Biodistribution of filamentous phage peptide libraries in mice. Mol. Biol. Rep. 2004, 31, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Wang, L.; Sha, R.; Liu, L.; Qian, J.; Ishimwe, N.; Zhang, W.; Qian, J.; Zhang, Y.; Wen, L. A blood circulation-prolonging peptide anchored biomimetic phage-platelet hybrid nanoparticle system for prolonged blood circulation and optimized anti-bacterial performance. Theranostics 2021, 11, 2278–2296. [Google Scholar] [CrossRef]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef] [PubMed]
- You, F.; Yin, G.; Pu, X.; Li, Y.; Hu, Y.; Huang, Z.; Liao, X.; Yao, Y.; Chen, X. Biopanning and characterization of peptides with fe3o4 nanoparticles-binding capability via phage display random peptide library technique. Colloids Surf. B Biointerfaces 2016, 141, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Ishikawa, Y.; Uraoka, Y.; Yamashita, I.; Yasueda, H. Selection of a novel peptide aptamer with high affinity for tio(2)-nanoparticle through a direct electroporation with tio(2)-binding phage complexes. J. Biosci. Bioeng. 2016, 122, 528–532. [Google Scholar] [CrossRef]
- Konoeda, H.; Takizawa, H.; Gower, A.; Zhao, M.; Adeyi, O.A.; Liu, M. Pharmacokinetics, tissue distribution and safety of gold nanoparticle/pkc delta inhibitor peptide hybrid in rats. Nanotoxicology 2020, 14, 341–354. [Google Scholar] [CrossRef]
- Kalishwaralal, K.; Luboshits, G.; Firer, M.A. Synthesis of gold nanoparticle: Peptide-drug conjugates for targeted drug delivery. Methods Mol. Biol. 2020, 2059, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, J.; Lei, J.; Ju, H. A core-shell nanoparticle-peptide@metal-organic framework as ph and enzyme dual-recognition switch for stepwise-responsive imaging in living cells. Chem. Commun. 2018, 54, 9155–9158. [Google Scholar] [CrossRef] [PubMed]
- Raju, C.V.; Manohara Reddy, Y.V.; Cho, C.H.; Shin, H.H.; Park, T.J.; Park, J.P. Highly sensitive electrochemical peptide-based biosensor for marine biotoxin detection using a bimetallic platinum and ruthenium nanoparticle-tethered metal-organic framework modified electrode. Food Chem. 2023, 428, 136811. [Google Scholar] [CrossRef]
- Bruun, T.U.J.; Andersson, A.C.; Draper, S.J.; Howarth, M. Engineering a rugged nanoscaffold to enhance plug-and-display vaccination. ACS Nano 2018, 12, 8855–8866. [Google Scholar] [CrossRef]
- Patwardhan, S.V.; Emami, F.S.; Berry, R.J.; Jones, S.E.; Naik, R.R.; Deschaume, O.; Heinz, H.; Perry, C.C. Chemistry of aqueous silica nanoparticle surfaces and the mechanism of selective peptide adsorption. J. Am. Chem. Soc. 2012, 134, 6244–6256. [Google Scholar] [CrossRef] [PubMed]
- Raja, I.S.; Kim, C.; Song, S.J.; Shin, Y.C.; Kang, M.S.; Hyon, S.H.; Oh, J.W.; Han, D.W. Virus-incorporated biomimetic nanocomposites for tissue regeneration. Nanomaterials 2019, 9, 1014. [Google Scholar] [CrossRef]
- Parent, K.N.; Deedas, C.T.; Egelman, E.H.; Casjens, S.R.; Baker, T.S.; Teschke, C.M. Stepwise molecular display utilizing icosahedral and helical complexes of phage coat and decoration proteins in the development of robust nanoscale display vehicles. Biomaterials 2012, 33, 5628–5637. [Google Scholar] [CrossRef]
- Zou, M.; Wang, J.; Shao, Z. Therapeutic potential of exosomes in tendon and tendon-bone healing: A systematic review of preclinical studies. J. Funct. Biomater. 2023, 14, 299. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Yu, Y. A review of recent advances in exosomes and allergic rhinitis. Front. Pharmacol. 2022, 13, 1096984. [Google Scholar] [CrossRef]
- Zheng, T. A review of the roles of specialized extracellular vesicles, migrasomes, and exosomes in normal cell physiology and disease. Med. Sci. Monit. 2023, 29, e940118. [Google Scholar] [CrossRef]
- Zheng, J.; Hu, X.; Zeng, Y.; Zhang, B.; Sun, Z.; Liu, X.; Zheng, W.; Chai, Y. Review of the advances in lipid anchors-based biosensors for the isolation and detection of exosomes. Anal. Chim. Acta 2023, 1263, 341319. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Han, X.; Li, C.; Sun, T.; Li, K.; Liu, X.; Liu, M. The status of industrialization and development of exosomes as a drug delivery system: A review. Front. Pharmacol. 2022, 13, 961127. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, M.; Ansari, M.N.M.; Razak, S.I.A.; Khan, M.U.A. A comprehensive review on the applications of exosomes and liposomes in regenerative medicine and tissue engineering. Polymers 2021, 13, 2529. [Google Scholar] [CrossRef]
- Mirzaaghasi, A.; Han, Y.; Ahn, S.H.; Choi, C.; Park, J.H. Biodistribution and pharmacokinectics of liposomes and exosomes in a mouse model of sepsis. Pharmaceutics 2021, 13, 427. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.; Ma, G.; Na, L.; Hudoklin, S.; Kreft, M.E.; Kostevsek, N.; Al-Jamal, W.T. Encapsulation of doxorubicin prodrug in heat-triggered liposomes overcomes off-target activation for advanced prostate cancer therapy. Acta Biomater. 2022, 140, 530–546. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Rombel, I.T.; Samli, K.N.; Zhou, X.; Brown, K.C. Isolation of multiple cell-binding ligands from different phage displayed-peptide libraries. Biosens. Bioelectron. 2006, 21, 1867–1875. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.P.; Li, S.; Brown, K.C. From phage display to nanoparticle delivery: Functionalizing liposomes with multivalent peptides improves targeting to a cancer biomarker. Bioconjug. Chem. 2013, 24, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Petrenko, V.A.; Torchilin, V.P. Optimization of landscape phage fusion protein-modified polymeric peg-pe micelles for improved breast cancer cell targeting. J. Nanomed. Nanotechnol. 2012, S4, 008. [Google Scholar] [CrossRef]
- Wang, T.; Kulkarni, N.; D’Souza, G.G.; Petrenko, V.A.; Torchilin, V.P. On the mechanism of targeting of phage fusion protein-modified nanocarriers: Only the binding peptide sequence matters. Mol. Pharm. 2011, 8, 1720–1728. [Google Scholar] [CrossRef]
- Wang, T.; Hartner, W.C.; Gillespie, J.W.; Praveen, K.P.; Yang, S.; Mei, L.A.; Petrenko, V.A.; Torchilin, V.P. Enhanced tumor delivery and antitumor activity in vivo of liposomal doxorubicin modified with mcf-7-specific phage fusion protein. Nanomedicine 2014, 10, 421–430. [Google Scholar] [CrossRef]
- Bedi, D.; Musacchio, T.; Fagbohun, O.A.; Gillespie, J.W.; Deinnocentes, P.; Bird, R.C.; Bookbinder, L.; Torchilin, V.P.; Petrenko, V.A. Delivery of sirna into breast cancer cells via phage fusion protein-targeted liposomes. Nanomedicine 2011, 7, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; D’Souza, G.G.; Bedi, D.; Fagbohun, O.A.; Potturi, L.P.; Papahadjopoulos-Sternberg, B.; Petrenko, V.A.; Torchilin, V.P. Enhanced binding and killing of target tumor cells by drug-loaded liposomes modified with tumor-specific phage fusion coat protein. Nanomedicine 2010, 5, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Meibohm, B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin. Pharmacokinet. 2013, 52, 855–868. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Latham, P.W. Therapeutic peptides revisited. Nat. Biotechnol. 1999, 17, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.K.; Viswanathan, M.; Kent, R.B.; Wood, C.R. Therapeutic peptides: Technological advances driving peptides into development. Curr. Opin. Biotechnol. 2006, 17, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H. Pharmacokinetics of biotech drugs: Peptides, proteins and monoclonal antibodies. Curr. Drug Metab. 2009, 10, 661–691. [Google Scholar] [CrossRef] [PubMed]
- Hosseinimehr, S.J.; Tolmachev, V.; Orlova, A. Liver uptake of radiolabeled targeting proteins and peptides: Considerations for targeting peptide conjugate design. Drug Discov. Today 2012, 17, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.E.; Fischer, V.; Connors, S.; Fisher, R.L.; Baldeck, J.P.; Maurer, G.; Brendel, K. Cyclosporin a metabolism in human liver, kidney, and intestine slices. Comparison to rat and dog slices and human cell lines. Drug Metab. Dispos. 1992, 20, 802–809. [Google Scholar]
- Peletier, L.A.; Gabrielsson, J. Dynamics of target-mediated drug disposition. Eur. J. Pharm. Sci. 2009, 38, 445–464. [Google Scholar] [CrossRef]
- Mager, D.E. Target-mediated drug disposition and dynamics. Biochem. Pharmacol. 2006, 72, 1–10. [Google Scholar] [CrossRef]
- Woo, S.; Krzyzanski, W.; Duliege, A.M.; Stead, R.B.; Jusko, W.J. Population pharmacokinetics and pharmacodynamics of peptidic erythropoiesis receptor agonist (era) in healthy volunteers. J. Clin. Pharmacol. 2008, 48, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Krzyzanski, W.; Doshi, S.; Xiao, J.J.; Perez-Ruixo, J.J.; Chow, A.T. Pharmacodynamics-mediated drug disposition (pdmdd) and precursor pool lifespan model for single dose of romiplostim in healthy subjects. AAPS J. 2010, 12, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Samtani, M.N.; Perez-Ruixo, J.J.; Brown, K.H.; Cerneus, D.; Molloy, C.J. Pharmacokinetic and pharmacodynamic modeling of pegylated thrombopoietin mimetic peptide (peg-tpom) after single intravenous dose administration in healthy subjects. J. Clin. Pharmacol. 2009, 49, 336–350. [Google Scholar] [CrossRef] [PubMed]
- Chirmule, N.; Jawa, V.; Meibohm, B. Immunogenicity to therapeutic proteins: Impact on pk/pd and efficacy. AAPS J. 2012, 14, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.F.; Bhansali, S.G.; Morris, M.E. Mechanistic determinants of biotherapeutics absorption following sc administration. AAPS J. 2012, 14, 559–570. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chu, T.; Wang, Y.; Wang, X. Radiolabeling and biodistribution of a nasopharyngeal carcinoma-targeting peptide identified by in vivo phage display. Acta Biochim. Biophys. Sin. 2007, 39, 624–632. [Google Scholar] [CrossRef]
- Yip, Y.L.; Hawkins, N.J.; Smith, G.; Ward, R.L. Biodistribution of filamentous phage-fab in nude mice. J. Immunol. Methods 1999, 225, 171–178. [Google Scholar] [CrossRef]
- Molenaar, T.J.; Michon, I.; de Haas, S.A.; van Berkel, T.J.; Kuiper, J.; Biessen, E.A. Uptake and processing of modified bacteriophage m13 in mice: Implications for phage display. Virology 2002, 293, 182–191. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Nagy, J.A.; Dvorak, J.T.; Dvorak, A.M. Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am. J. Pathol. 1988, 133, 95–109. [Google Scholar] [PubMed]
- Wiseman, R.L.; Berkowitz, S.A.; Day, L.A. Different arrangements of protein subunits and single-stranded circular DNA in the filamentous bacterial viruses fd and pf1. J. Mol. Biol. 1976, 102, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Hagedorn, H.; Heuck, C.C.; Hinrichsen, M.; Ludwig, H. The ionic properties of the filamentous bacteriophages pf1 and fd. J. Biol. Chem. 1986, 261, 1653–1655. [Google Scholar] [CrossRef]
- Khalil, A.S.; Ferrer, J.M.; Brau, R.R.; Kottmann, S.T.; Noren, C.J.; Lang, M.J.; Belcher, A.M. Single m13 bacteriophage tethering and stretching. Proc. Natl. Acad. Sci. USA 2007, 104, 4892–4897. [Google Scholar] [CrossRef]
- Driessen, W.H.; Bronk, L.F.; Edwards, J.K.; Proneth, B.; Souza, G.R.; Decuzzi, P.; Pasqualini, R.; Arap, W. On the synergistic effects of ligand-mediated and phage-intrinsic properties during in vivo selection. Adv. Genet. 2010, 69, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Naderi, S.; Pouget, E.; Ballesta, P.; van der Schoot, P.; Lettinga, M.P.; Grelet, E. Fractional hoppinglike motion in columnar mesophases of semiflexible rodlike particles. Phys. Rev. Lett. 2013, 111, 037801. [Google Scholar] [CrossRef] [PubMed]
- Merkel, R.; Nassoy, P.; Leung, A.; Ritchie, K.; Evans, E. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature 1999, 397, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.A.; Calderwood, D.A. Forces and bond dynamics in cell adhesion. Science 2007, 316, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Decuzzi, P.; Ferrari, M. The receptor-mediated endocytosis of nonspherical particles. Biophys. J. 2008, 94, 3790–3797. [Google Scholar] [CrossRef]
- Ivanenkov, V.; Felici, F.; Menon, A.G. Uptake and intracellular fate of phage display vectors in mammalian cells. Biochim. Biophys. Acta 1999, 1448, 450–462. [Google Scholar] [CrossRef]
- Ziegler, A.; Blatter, X.L.; Seelig, A.; Seelig, J. Protein transduction domains of hiv-1 and siv tat interact with charged lipid vesicles. Binding mechanism and thermodynamic analysis. Biochemistry 2003, 42, 9185–9194. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, E.; Kitas, E.; Seelig, J. Binding of oligoarginine to membrane lipids and heparan sulfate: Structural and thermodynamic characterization of a cell-penetrating peptide. Biochemistry 2005, 44, 2692–2702. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Dabrowska, K. Phage therapy: What factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med. Res. Rev. 2019, 39, 2000–2025. [Google Scholar] [CrossRef]
- Park, H.; Sut, T.N.; Yoon, B.K.; Zhdanov, V.P.; Kim, J.W.; Cho, N.J.; Jackman, J.A. Multivalency-induced shape deformation of nanoscale lipid vesicles: Size-dependent membrane bending effects. J. Phys. Chem. Lett. 2022, 13, 1480–1488. [Google Scholar] [CrossRef]
- Levitan, B. Stochastic modeling and optimization of phage display. J. Mol. Biol. 1998, 277, 893–916. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.; Mitragotri, S. Delivery of sirna and other macromolecules into skin and cells using a peptide enhancer. Proc. Natl. Acad. Sci. USA 2011, 108, 15816–15821. [Google Scholar] [CrossRef]
- Chen, Y.; Shen, Y.; Guo, X.; Zhang, C.; Yang, W.; Ma, M.; Liu, S.; Zhang, M.; Wen, L.P. Transdermal protein delivery by a coadministered peptide identified via phage display. Nat. Biotechnol. 2006, 24, 455–460. [Google Scholar] [CrossRef]
- Gurung, S.; Khan, F.; Gunassekaran, G.R.; Yoo, J.D.; Poongkavithai Vadevoo, S.M.; Permpoon, U.; Kim, S.H.; Kim, H.J.; Kim, I.S.; Han, H.; et al. Phage display-identified pd-l1-binding peptides reinvigorate t-cell activity and inhibit tumor progression. Biomaterials 2020, 247, 119984. [Google Scholar] [CrossRef]
- Wan, X.M.; Chen, Y.P.; Xu, W.R.; Yang, W.J.; Wen, L.P. Identification of nose-to-brain homing peptide through phage display. Peptides 2009, 30, 343–350. [Google Scholar] [CrossRef]
- Zahid, M.; Phillips, B.E.; Albers, S.M.; Giannoukakis, N.; Watkins, S.C.; Robbins, P.D. Identification of a cardiac specific protein transduction domain by in vivo biopanning using a m13 phage peptide display library in mice. PLoS ONE 2010, 5, e12252. [Google Scholar] [CrossRef] [PubMed]
- Kanki, S.; Jaalouk, D.E.; Lee, S.; Yu, A.Y.; Gannon, J.; Lee, R.T. Identification of targeting peptides for ischemic myocardium by in vivo phage display. J. Mol. Cell. Cardiol. 2011, 50, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Rajotte, D.; Ruoslahti, E. Membrane dipeptidase is the receptor for a lung-targeting peptide identified by in vivo phage display. J. Biol. Chem. 1999, 274, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.J.; Smith, M.W.; Griffiths, P.C.; McKeown, N.B.; Gumbleton, M. Enhanced pulmonary absorption of a macromolecule through coupling to a sequence-specific phage display-derived peptide. J. Control Release 2011, 151, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Lee, J.H.; Chung, H.K.; Ju, E.J.; Song, S.Y.; Jeong, S.Y.; Choi, E.K. Application of peptide displaying phage as a novel diagnostic probe for human lung adenocarcinoma. Amino Acids 2016, 48, 1079–1086. [Google Scholar] [CrossRef]
- Koivistoinen, A.; Ilonen, I.I.; Punakivi, K.; Rasanen, J.V.; Helin, H.; Sihvo, E.I.; Bergman, M.; Salo, J.A. A novel peptide (thx) homing to non-small cell lung cancer identified by ex vivo phage display. Clin. Transl. Oncol. 2013, 15, 492–498. [Google Scholar] [CrossRef]
- Haque, M.E.; Khan, F.; Chi, L.; Gurung, S.; Vadevoo, S.M.P.; Park, R.W.; Kim, D.K.; Kim, S.K.; Lee, B. A phage display-identified peptide selectively binds to kidney injury molecule-1 (kim-1) and detects kim-1-overexpressing tumors in vivo. Cancer Res. Treat. 2019, 51, 861–875. [Google Scholar] [CrossRef]
- Costantini, T.W.; Eliceiri, B.P.; Putnam, J.G.; Bansal, V.; Baird, A.; Coimbra, R. Intravenous phage display identifies peptide sequences that target the burn-injured intestine. Peptides 2012, 38, 94–99. [Google Scholar] [CrossRef]
- Duerr, D.M.; White, S.J.; Schluesener, H.J. Identification of peptide sequences that induce the transport of phage across the gastrointestinal mucosal barrier. J. Virol. Methods 2004, 116, 177–180. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Zhang, Y.; Hu, Z.; Hu, D.; Pan, Y.; Ou, S.; Liu, G.; Yin, X.; Zhao, J.; et al. Panning and identification of a colon tumor binding peptide from a phage display peptide library. J. Biomol. Screen. 2007, 12, 429–435. [Google Scholar] [CrossRef]
- Bai, F.; Liang, J.; Wang, J.; Shi, Y.; Zhang, K.; Liang, S.; Hong, L.; Zhai, H.; Lu, Y.; Han, Y.; et al. Inhibitory effects of a specific phage-displayed peptide on high peritoneal metastasis of gastric cancer. J. Mol. Med. 2007, 85, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yan, J.; Chen, X.; Wei, J.; Yu, L.; Liu, F.; Li, L.; Liu, B. Identification of a peptide binding to cancer antigen kita-kyushu lung cancer antigen 1 from a phage-display library. Cancer Sci. 2021, 112, 4335–4345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Y.; Wang, J.; Zhang, Y.; Chen, J.; Pan, Y.; Ren, L.; Hu, Z.; Zhao, J.; Liao, M.; et al. Screening and identification of a targeting peptide to hepatocarcinoma from a phage display peptide library. Mol. Med. 2007, 13, 246–254. [Google Scholar] [CrossRef]
- Joyce, J.A.; Laakkonen, P.; Bernasconi, M.; Bergers, G.; Ruoslahti, E.; Hanahan, D. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2003, 4, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zheng, L.; Wu, K.; Zhang, B. Identification and validation of a new peptide targeting pancreatic beta cells. Molecules 2022, 27, 2286. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Wang, J.; Yang, L.; Zhao, Z.; Lu, K.; Zhao, L.; Xiao, J.; Li, Z.; Shi, Z. Targeting osteosarcoma vasculature with peptide obtained by phage display. Contemp. Oncol. 2014, 18, 165–170. [Google Scholar] [CrossRef]
- Sun, X.; Niu, G.; Yan, Y.; Yang, M.; Chen, K.; Ma, Y.; Chan, N.; Shen, B.; Chen, X. Phage display-derived peptides for osteosarcoma imaging. Clin. Cancer Res. 2010, 16, 4268–4277. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Dooner, M.S.; Valinski, H.M.; Mihaliak, A.M.; Quesenberry, P.J.; Becker, P.S. A specific heptapeptide from a phage display peptide library homes to bone marrow and binds to primitive hematopoietic stem cells. Stem Cells 2004, 22, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Kim, H.S.; Kim, K.H.; Kim, E.B.; Cho, C.S.; Kang, S.K.; Choi, Y.J. Identification of a novel peptide ligand targeting visceral adipose tissue via transdermal route by in vivo phage display. J. Drug Target. 2011, 19, 805–813. [Google Scholar] [CrossRef]
- Zapi-Colin, L.A.; Gutierrez-Gonzalez, G.; Rodriguez-Martinez, S.; Cancino-Diaz, J.C.; Mendez-Tenorio, A.; Perez-Tapia, S.M.; Gomez-Chavez, F.; Cedillo-Pelaez, C.; Cancino-Diaz, M.E. A peptide derived from phage-display limits psoriasis-like lesions in mice. Heliyon 2020, 6, e04162. [Google Scholar] [CrossRef]


{kind=link}
| Vector Type | Coat Protein | Displayed Molecules (n) | Comments |
|---|---|---|---|
| 3 | pIII | 5 | The type 3 vectors carry only one modified pIII gene. |
| 33 | pIII | 1–3 | The type 33 vector systems carry both a wild-type pIII phage gene and a modified pIII gene. |
| 3 + 3 | pIII | ~1 | The vector systems referred to as 3 + 3 have a modified pIII gene on a phagemid and utilize helper phage to introduce an additional wild-type pIII gene. |
| 8 | pVIII | 2700 | The type 8 vectors carry only one modified pVIII gene. |
| 88 | pVIII | ~100–200 | The type 88 vector systems carry both the wild-type pVIII phage gene and a modified pVIII gene. |
| 8 + 8 | pVIII | ~100–200 | The 8 + 8 vector systems have a modified pVIII gene on a phagemid and utilize helper phage to introduce an additional wild-type pVIII gene. |
| Strategy | Targets | Advantages | Disadvantages |
|---|---|---|---|
| In vitro | Recombinant proteins and other isolated biomolecules. | Easy setup, fast, cheap, low Kd. | Poor pharmacokinetics, low specificity, low stability. |
| In situ | Cell lines, organoids, ex vivo tissues and organs. | Reflects the complex milieu of a cell, low-to-mid range Kd, enhanced binding specificity and stability. | Higher Kd compared to in vitro selections. |
| In vivo | Live animals and patients. | Reflects the complexity of an in vivo milieu, enhanced pharmacokinetics, binding specificity, and stability. | Mid-range Kd, expensive, time-consuming. |
| Tissue/Organ | Name | Target | Effect | Sequence | Reference |
|---|---|---|---|---|---|
| Cancer-Immune Interactions | PD-L1 Pep-1 PD-L1 Pep-2 | Programed Cell Death Ligand 1 (PD-L1) | Tumor Homing/ Blocking of T cell Function | CLQKTPKQC CVRARTR | [164] |
| Peripheral Blood Cells | BCP-1 BCP-2 | RGD Integrin Targeting on Peripheral Blood Cells | Prolong Circulation within Blood | CNARGDMHC CIVRGDNVC | [84] |
| Blood-Brain Barrier | Clone 7 | Unknown | Nose to Brain Translocation Capability/ Olfactory and Brain Homing | ACTTPHAWLCG | [165] |
| Heart | Cardiac Targeting Peptide (CTP) | Unknown | Heart/Cardio Myoblast Homing and Internalization | APWHLSSQYSRT | [166] |
| Ischemic Heart | None | Unknown | Myocardium Damaged by Ischemia- Reperfusion | CSTSMLKAC | [167] |
| Lung | GFE-1 | Membrane Dipeptidase (MDP) | Mouse Lung Vasculature | CGFECVRQCPERC | [168] |
| Lung Epithelial | LTP-1 | Unknown | Pulmonary Epithelial Translocation | CTSGTHPRC | [169] |
| Lung Cancer | Pep1 | Unknown | Lung Adenocarcinoma with Increased Uptake Post Radiation Treatment | CAKATCPAC | [170] |
| Non-Small Cell Lung Cancer | Thx | Unknown | Non-Small Cell Lung Cancer | ARRPKLD | [171] |
| Kidney/Kidney Cancer | None | Kidney Injury Molecule (KIM-1) | Kidney Cancer | CNWMINKEC | [172] |
| Kidney | RCC1-02 | Unknown | Redistribution Towards Kidney Clearance/ Avoidance of Protein Reabsorption | AGGLSFGTRRFEIGR | [78] |
| Intestine | 4–1 4–11 | Unknown | Internalization by Normal Intestine | SGHQLLLNKMP SFKPSGLPAQSL | [173] |
| Intestine | Sequence Homology (HIV gp120) | Translocation Across the Intestine | YPRLLTP | [174] | |
| Injured Intestine | 4–5 | Unknown | Internalized by Injured Intestine | ILANDLTAPGPR | [173] |
| Colon Cancer | CP15 | Unknown | Colon Cancer | VHLGYAT | [175] |
| Peritoneal Metastasis of Gastric Cancer | pIII | Unknown | Homing to-and Prevention of- Metastases of Gastric Cancer | SMSIASPYIALE | [176] |
| Gastric Cancer | Peptide 1131 | Kita-Kyushu Lung Cancer Antigen-1 (KK-LC-1) | Gastric Cancer | Not given | [177] |
| Liver Cancer | HCBP1 | Unknown | Liver Cancer | FQHPSFI | [178] |
| Pancreas | None | Putative EphA4 Receptor/ Sequence Similarity with Ephrin-A Ligands | Pancreas Islet Cells | CHVLWSTRC CVSNPRWKC | [15] |
| Pancreatic cancer | RGR RSR KAA KAR VGV EYQ | Sequence Similarity with Various Proteins | Angiogenic Vasculature of Pancreatic Islet Tumorigenesis | CRGRRST CRSRKG CKAAKNK FRVGVADV CEYQLDVE | [179] |
| Pancreas | None | Unknown | Pancreatic Beta Cells | LNTPLKS | [180] |
| Bone Cancer | NF-1 | Unknown | Osteosarcoma Vasculature | CTKPDKGYC | [181] |
| Bone Cancer | OSP-1 | Putative Heparan Sulfate Proteoglycan | Osteosarcoma | ASGALSPSRLDT | [182] |
| Bone Marrow | None | Putative Sequence Similarity with CD84 | Bone Marrow | STFTKSP | [183] |
| Adipose | TDA1 | Unknown | Transdermal Targeting of Visceral Adipose Tissue | CGLHPAPQC | [184] |
| Skin/psoriatic lesions | Pep3D | Interferon- Alpha Receptor | Reduces Psoriasis Symptoms | CIGNSNTLC | [185] |
| Skin | T2 Peptide | Lipids | Skin Penetrating | LVGVFH | [92] |
| Skin | None | Unknown | Transient Pore Formation Within the Skin for Transdermal Protein Delivery | ACSSSPSKHCG | [163] |
| Skin | Skin Penetrating and Cell Entering (SPACE) Peptide | Unknown | Keratinocytes, Fibroblasts, and Endothelial Cells | ACTGSTQHQCG | [162] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asar, M.; Newton-Northup, J.; Soendergaard, M. Improving Pharmacokinetics of Peptides Using Phage Display. Viruses 2024, 16, 570. https://doi.org/10.3390/v16040570
Asar M, Newton-Northup J, Soendergaard M. Improving Pharmacokinetics of Peptides Using Phage Display. Viruses. 2024; 16(4):570. https://doi.org/10.3390/v16040570
Chicago/Turabian StyleAsar, Mallika, Jessica Newton-Northup, and Mette Soendergaard. 2024. "Improving Pharmacokinetics of Peptides Using Phage Display" Viruses 16, no. 4: 570. https://doi.org/10.3390/v16040570