1. Introduction
The
Flaviviridae family encompasses over 80 species of small, enveloped, single-stranded RNA viruses classified into four genera: orthoflavivirus, pestivirus, pegivirus, and hepacivirus [
1,
2]. These viruses are usually host-specific and mainly infect birds and mammals. Horses are susceptible to several flaviviruses, some of which are relevant zoonotic agents [
1]. In the last decade, a recently discovered virus, the equine hepacivirus (hereon EqHV; family:
Flaviviridae; genus: hepacivirus), initially described as non-primate hepacivirus in 2012 [
3], then classified as hepacivirus A in 2016 [
4], and recently renamed hepacivirus equi [
2], gained greater attention due to its high genetic homology with the human hepatitis C virus (HCV) [
3] and due to its global diffusion. As the virus is present worldwide, several studies focused on its transmission routes, which remain elusive and subject of scientific debate: to date, only parenteral transmission [
5,
6,
7,
8] and sporadic cases of vertical transmission [
9,
10] have been successfully demonstrated, while more data are required to evaluate the potential role of insects [
11] and the role of the proximity of individuals in horizontal transmission [
9,
12,
13,
14]. Sexual transmission has not been investigated yet. Noteworthy is that the virus was also found in commercial equine products (serum and plasma) [
3,
4,
5,
6,
15,
16,
17,
18], meaning that it could evade sterilizing and control procedures, increasing the risk of its spread by veterinary therapeutic treatments [
19,
20]. The onset of the EqHV infection is relatively rapid: viral RNA can be detected by PCR one week post-infection in the absence of clinical signs [
5,
21], with a high chance of spreading unnoticed within a holding during the first stages of the infection [
22]. The outcome of EqHV infection is either a sub-clinical hepatitis, which usually self-clears in a few weeks [
23], or a persistent infection which could last up to a year [
9,
24,
25,
26,
27]. Infected individuals usually show mild signs to none, but there are reports of poor performance, jaundice, fatigue, or discomfort in viremic subjects [
14,
19,
24,
27,
28]. Moreover, correlations between EqHV infection and an increase in specific liver enzymes were assessed [
3,
5,
7,
8,
21,
23,
24,
27,
28]; however, these values are also reported to frequently remain within the reference intervals or mildly increase in viremic animals [
8,
21,
29,
30]. Correlation patterns between EqHV infection and a selection of horse characteristics (age, breed, sex, production category) were also investigated. Young horses (<8 years) are often more susceptible [
9,
31,
32,
33,
34,
35,
36] compared to older horses (>10 years) [
14,
37]. In thoroughbreds, a higher percentage of PCR positivity was described when compared to other breeds [
15,
21,
31,
32,
33,
34,
38]; females seem to be more prone to infection [
35,
36,
37] compared to males [
30], and competition horses show similar higher trends of susceptibility [
31,
39,
40,
41]. These data which, however, require further verification, hint that there could be implicit management practices exposing certain individuals to a higher risk of infection. In recent years, several studies have assessed EqHV biomolecular and serological prevalence in Africa [
34,
36], in North America [
3,
20], in South America [
30,
35,
39], in Asia [
14,
26,
31,
32,
37,
40,
41,
42,
43,
44], and in Australia [
45]. Overall, the worldwide biomolecular EqHV prevalence ranges from <1% to 18.2% and the serological prevalence ranges from 23.9% to 83.7% (taking into account studies which include at least N ≥ 100 subjects, sampled and examined within the same time span). In Europe, EqHV prevalence has been thoroughly investigated: the first report was in 2012, in the UK [
29], which highlighted a biomolecular prevalence of 2.1% (3/143); in Germany, higher biomolecular prevalence was reported, ranging from 2.4% to 18.2% [
15,
21,
33,
46,
47]; in Austria, a prevalence range from 0.38% to 4.15% was reported [
11]; while in France the range was from 5.6% to 6.2% [
25,
38]. In Italy, the only study estimating EqHV prevalence was published in 2017 [
48], reporting a biomolecular prevalence of 4.7% in horses (91/1932), with the sampling performed only in two restricted Italian geographical areas: the north east and the south east, leaving out most of the peninsula and the islands.
The aims of the present study were: estimate EqHV national biomolecular prevalence in horses; verify if any statistical differences exist among the prevalences estimated for production categories (competition; equestrian; work and meat; reproduction) and macro-regions of Italy (north, center, south, and islands); perform a phylogenetic analysis on the sequences obtained from the positive samples; assess the mutation level of the analyzed sequences and also investigate their effect on the NS3 protein tertiary structure.
4. Discussion
Here we present the first study on EqHV prevalence at a national level, with the aim of providing further data regarding the distribution of this virus in Italy, both geographically and among horse production categories. In addition, we present the results of the phylogenetic and the NS3 tertiary structure analysis.
The study presents some limitations: although it was designed as a cross-sectional study, the COVID-19 pandemic in 2020 brought activities almost to a complete halt. This obliged us to further extend the sampling time for over a year and a half to reach the required sample size. Thus, the prevalence data presented cannot be considered as punctual, but instead refer to a time span. Furthermore, the study was designed to proportionally sample the horse population, reflecting its distribution on Italian territory, in order to estimate an accurate EqHV biomolecular prevalence in Italian horse production categories between 2019–2022. However, the collection of samples from some provinces and regions was harder during and after the COVID-19 pandemic; nevertheless, the sampling level achieved was considered reliable since the SE resulted in <5%, as set in the study design. Slight SE variances were observed in the macro-region comparison, but all values were close to the expected one. Another limitation of the present study was that data on the age, breed, and sex of the analyzed horses were not collected and therefore no retrospective comparisons, nor statistical analysis could be inferred regarding the influence that these risk factors could have played in the study scenario. Investigating these variables would require a greater number of samples that was not achievable with the funding resources available for the project. For the same reason, prevalence in donkeys and mules was not investigated, and priority was given to horses considering that they are the species most present and with the highest economic value.
However, despite these limitations, this study presents the first datum of EqHV biomolecular prevalence at national level in Italian horses. The described prevalence of 4.27% is coherent with what was previously reported in Italy (4.7%) [
48] and within the range of biomolecular prevalence reported both in Europe (<1–18.2%) [
11,
15,
21,
25,
29,
33,
38,
46,
47,
57,
58] and worldwide (3.2–46.2%) [
3,
14,
20,
26,
30,
31,
32,
34,
35,
36,
37,
39,
40,
41,
42,
43,
44,
45].
It should be pointed out that higher biomolecular prevalence (40–46.2%) is described in the literature [
22,
26,
33,
41] when the sample is collected within the same holdings; on the contrary, lower prevalence (3.4–5.6%) is described when the sampling was set to assess the prevalence in a geographic area [
11,
25,
40,
48]. Intra-premises prevalence appears higher worldwide; in fact, once introduced in a stable, the virus seems to spread rapidly with a still unclear transmission route and, because of its asymptomatic development, it is feasible that one or more carriers can infect other horses without showing any evident clinical signs.
The sampling was designed to also investigate four horse production categories: equestrian, competition, work/meat, and reproduction, which reflect the different categories within which holdings are officially classified by the Italian animal health authorities. This categorization was established to study whether different management practices could play a role in the spread of the virus. As a matter of fact, the literature reports that competition horses and breeding horses are often more susceptible to infection than other categories [
31,
35,
36,
37,
39,
40,
41]. In this study, the majority of PCR-positive samples were grouped within the competition and reproduction categories, although statistical analysis did not highlight any significant difference among the production categories. Further studies are necessary to confirm these results with a higher number of samples.
Nonetheless, the data collected in this study can be useful to highlight and confirm that EqHV infection poses a potential threat to animal health, being spread across all categories, and could have a tangible impact on the sport industry, which has the highest economic turnover. In fact, poor performances, lethargy, apathy and fatigue are widely reported [
14,
19,
24,
27,
28,
33], with occasional cases of euthanasia due to poor overall health conditions and severe hepatic damage [
19,
21,
24]. Data on EqHV biomolecular prevalence also highlight another issue related to therapies using blood, plasma, and other blood derivatives that are commercially available, as well as blood transfusions between horses. EqHV was already detected in some commercial blood products [
15,
16,
17,
18], and therefore it is crucial to amend quality control analysis to certify the commercial products as EqHV-free (together with the exclusion of other etiological agents that can be relevant for equine health), and to include this virus in veterinary diagnostic protocols to control its spread in the equine population via non-commercial transfusions.
At the present time there is no therapy available for EqHV, thus biosecurity measures to reduce the spread of the disease on a holding and between holdings are as follows: isolate the suspect case(s), suspend any production of blood products (if on a holding with this purpose), and periodically control the animals to monitor for the presence of the virus. Infection may last for a period of six months [
5,
6,
8] after which the subject recovers, and no natural cases of resurgence of infection are reported so far, although re-infection has been reported [
7,
8]. On the contrary, if the virus can still be detected after six months, the subject is considered chronically infected [
5,
6,
24,
26,
27,
28,
59] and should be managed with caution.
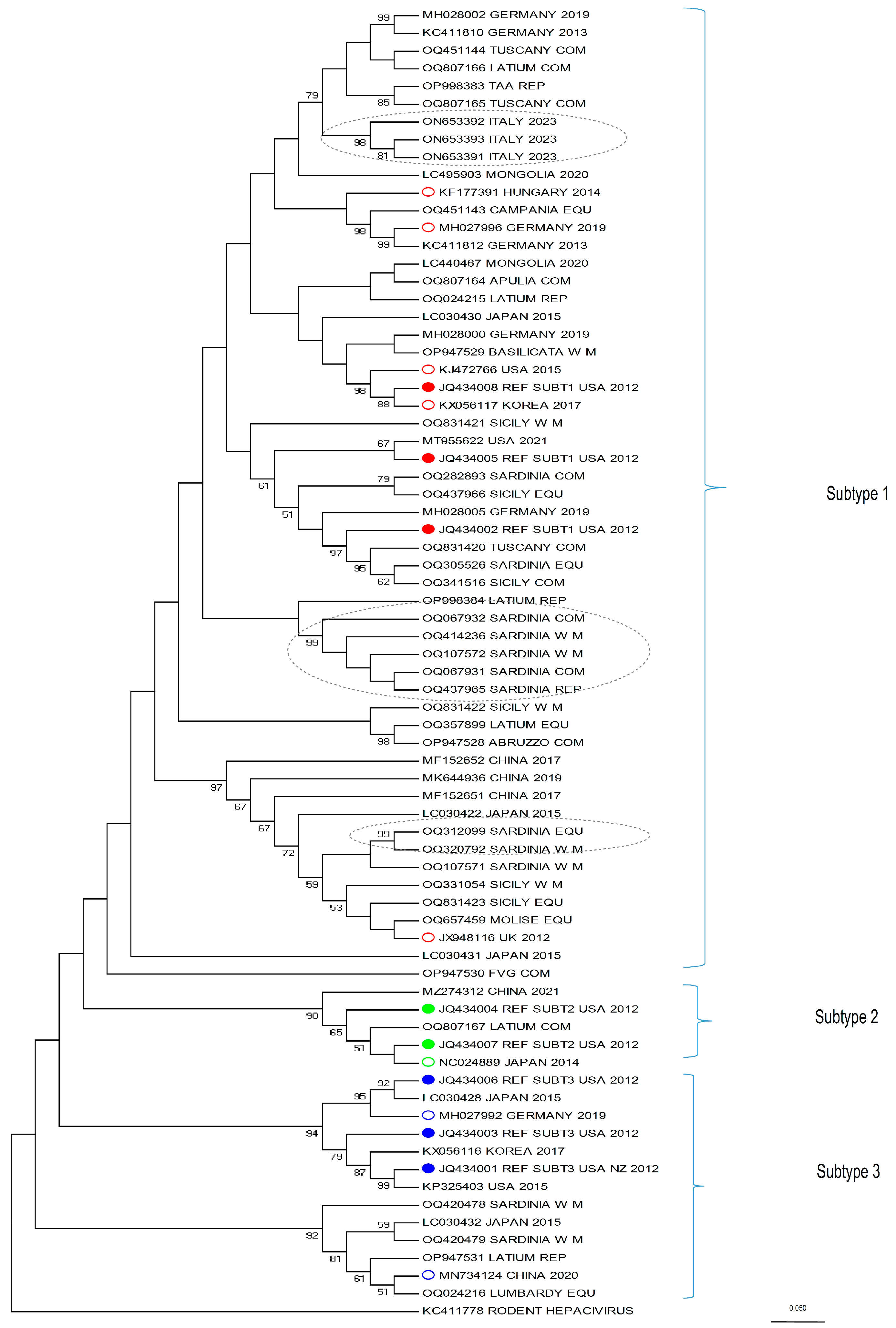
This study also provides comprehensive data on the phylogenetic characteristics of the isolates from the whole Italian territory, integrating the already available data that were limited to some regions only. For this purpose, we analyzed a portion of the NS3 fragment, which is the serine protease/helicase domain of the viral polyprotein. Although EqHV is an RNA virus, and therefore prone to mutation, the strains of the present study mostly belong to the three known EqHV sub-types (
Figure 2). As expected from the literature [
30,
37,
44], the majority of the sequences (30) clustered within sub-type 1, while four were identified as sub-type 3 and only one sequence as sub-type 2.
Some clusters in sub-type 1 (identified in
Figure 2 with grey dotted ellipses) are of particular interest. From top to bottom, the first cluster is represented by three very recent sequences from an Italian EqHV outbreak (kindly provided by the authors of [
22]), which are relevant because the authors could follow up the infection of a few horses for several months, allowing for the tracking of viral mutation within the same subjects. This is the case for ON653391 and ON653393, detected from the same horse sampled six months apart, which clearly shows that EqHV is prone to mutations (for more in-depth considerations, see [
22]). The second and third clusters, instead, include sequences from the present study and are located mid-way and at the bottom of sub-type 1, respectively. The first one includes five sequences from Sardinia (OQ067932, OQ414236, OQ107572, OQ067931, OQ437965) and the second includes two sequences, also from Sardinia (OQ312099 and OQ320792). Both clusters show a 99 bootstrap value. A retrospective tracing of the subjects allowed us to determine that (a) the premises of origin are distant from each other (>12 km to <250 km), apart from two samples which do not belong to the same cluster, nor to the same production category, but for which the stables of origin are located 2.1 km apart (Accession Number ID: OQ067932; OQ320792); (b) being geographically distributed, they could hardly share the same veterinarian, except from the aforementioned two subjects; (c) none of the seven horses had a history of movement to or from the other stables, according to the data available on the Veterinary Information System Online Database; (d) data on veterinary treatments (such as vaccines, plasma transfusion, etc.) could not be retrieved. The reason why these sequences cluster so closely together, then, cannot be assessed confidently, but it must be considered that each stable held a discrete number of horses (>1 to <21) at the time of sampling, therefore it cannot be ruled out that the movement of an EqHV-positive horse, different from those sampled, could be the source of infection for each respective cluster.
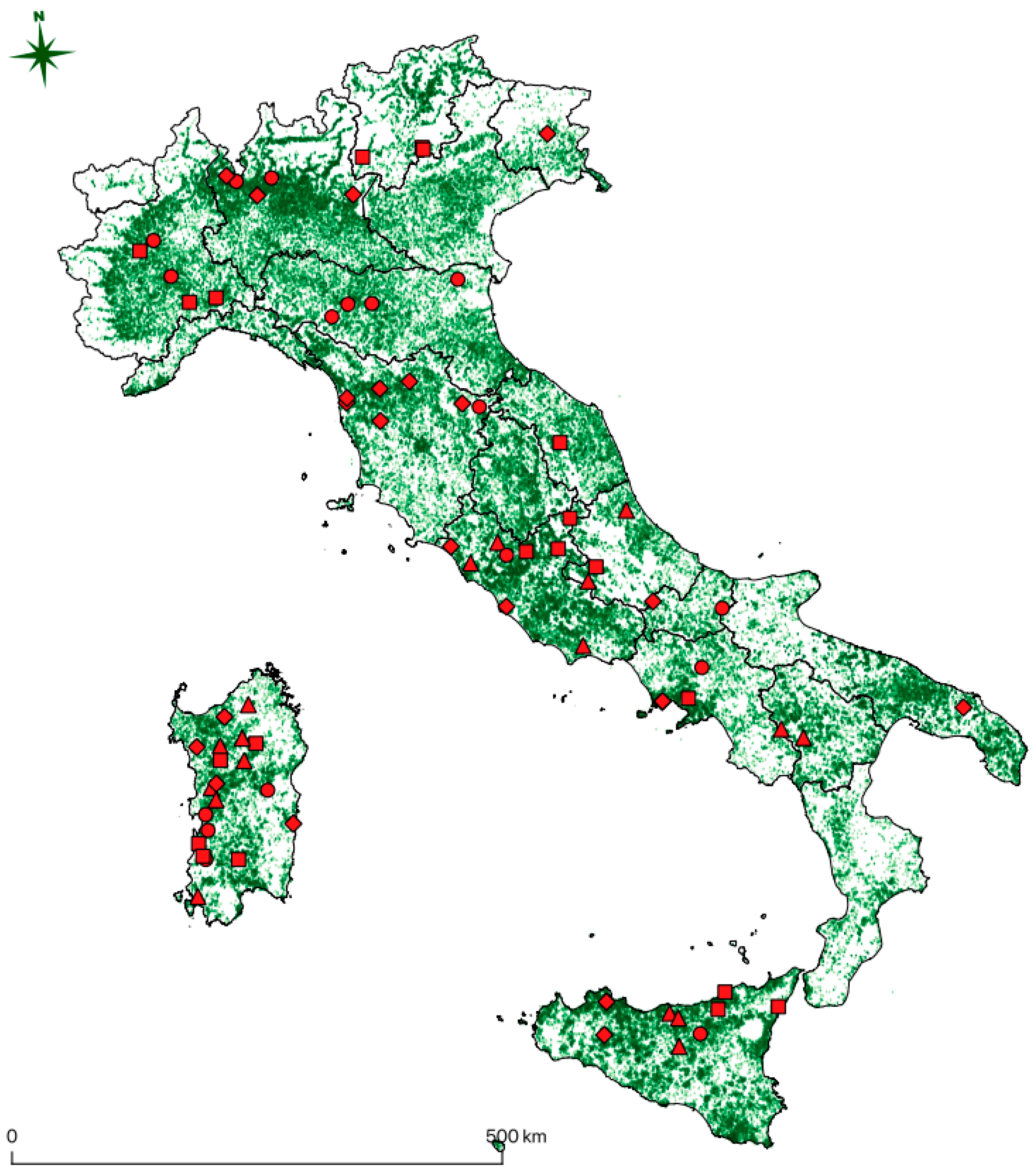
Almost all of the positive subjects detected in this study, as reported in
Figure 1, live in territory with a high density of equine premises, thus representing a threat for the spread of the virus through movement or other unknown transmission routes. Movements from one stable to another are indeed a common practice in the horse industry, either for trade, management, breeding, or sport activities. Since the virus appears widespread and the sequences are phylogenetically close regardless of their origin, presumably movements have played a role in the spread of the three sub-types worldwide in the last decade, and probably before that. When it comes to horse pre-movement sanitary controls, several diseases are screened, but EqHV is not currently present in routine laboratory diagnosis, at least in Italy, and probably also in most countries of the world. As preventive measures, stricter diagnostic protocols when moving animals should be considered, especially when susceptible breeds or valuable individuals are involved.
Further, regarding the phylogenetic analysis, another interesting aspect of the tree should be discussed: included in sub-type 3 there is a branch (the second branch bottom-up, identified by a 92 bootstrap value) which clusters alone and originates from a different node when compared to the other branches of the tree. Four sequences from the present study are included in this group (OQ420478, OQ420479, OP947531, OQ024216) along with two sequences retrieved from Genbank (LC030432 [
31] and MN734124 [
37]). Other recent phylogenetic analyses [
22,
26,
37,
44,
60] and the original paper from [
31], show a similar outcome. In these papers, in fact, the branch, identified either by LC030432 [
31], MN734124 [
37], or both, separates from the node which includes the sequences used as reference for sub-type 3 (JQ434001, JQ434003, JQ434006 [
3]). This suggests that these sequences could substantially differ from those included in sub-type 3 and from the other sub-types; it could be hypothesized that this branch was never part of EqHV sub-type 3, but it represents a totally different sub-type which was until now under-represented and that was therefore considered part of sub-type 3.
The most accredited criteria supporting the proposal of new sub-types for EqHV are those described in the literature for HCV [
4,
61,
62], which were already used to introduce the presence of sub-type 2 [
30,
38] and 3 [
41] for EqHV. These criteria identify the need for specific parameters to assess the detection of a new sub-type, such as a “complete or nearly complete coding region sequence differing from other sequences by at least 15% of nucleotide positions” and “sequence information from at least two other isolates in core/E1 […] and NS5B” [
62], which could not be fulfilled in the present study mainly for funding reasons. However, a larger set of sequences, representative of this branch and that could be used as reference, are those of the NS3 protein submitted by [
31] (LC030420, LC030425, LC030426, LC030427, LC030432), the complete polyprotein sequence by [
37] (MN734124), and the new partial NS3 sequences from the present study (OQ420478, OQ420479, OP947531, OQ024216). We are strongly looking forward to further studies and broader sampling from horses worldwide to verify this hypothesis that could update the classification of EqHV.
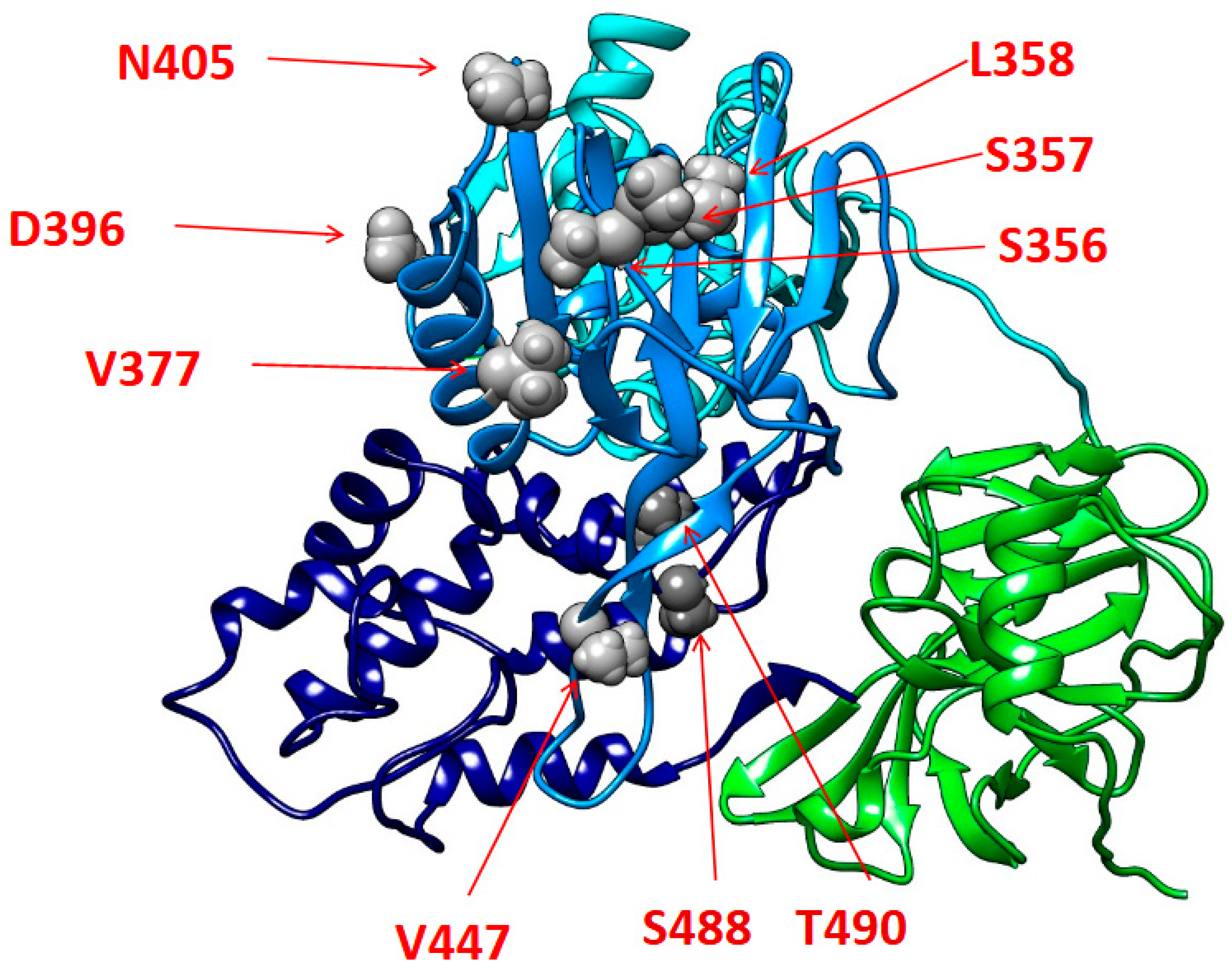
Regarding the mutational analysis, most of the identified mutations reside within the alfa-helixes of the II and III subdomains of the NS3 helicase functional regions. In particular, these mutations are localized in portions that are critical for the RNA binding and the following unwinding activity of NS3. In this regard, previous studies, in reference to the homologous NS3 protein of the human hepatitis C virus, have shown that amino acid substitutions in NS3 C-terminal alfa-helixes can influence RNA replication with a beneficial impact on the variant’s replicative fitness [
63]. On this basis, it could be speculated that the reported mutations may play a similar role, with a potential enhancing impact on EqHV replicative capability. However, further in-depth in vitro studies are necessary to confirm how the mutations accumulated in these regions could impact on NS3 proper folding and its binding affinity to viral RNA, and, in turn, to finally elucidate their potential implications on the viral replication activity. Lastly, a more extensive and comprehensive mutational analysis of full-length EqHV sequences will be critical to better characterize the mutational profiles of EqHV strains circulating in Italy.

 ,
, 



{kind=link}
{kind=link}
{kind=link}