
Co-Transcriptional Regulation of HBV Replication: RNA Quality Also Matters



{kind=link}
{kind=link}
Abstract
1. Introduction
2. cccDNA Transcription: A Key Step of HBV Replication
2.1. Generalities on cccDNA
2.2. cis-Element Controlling cccDNA Transcriptional Activity
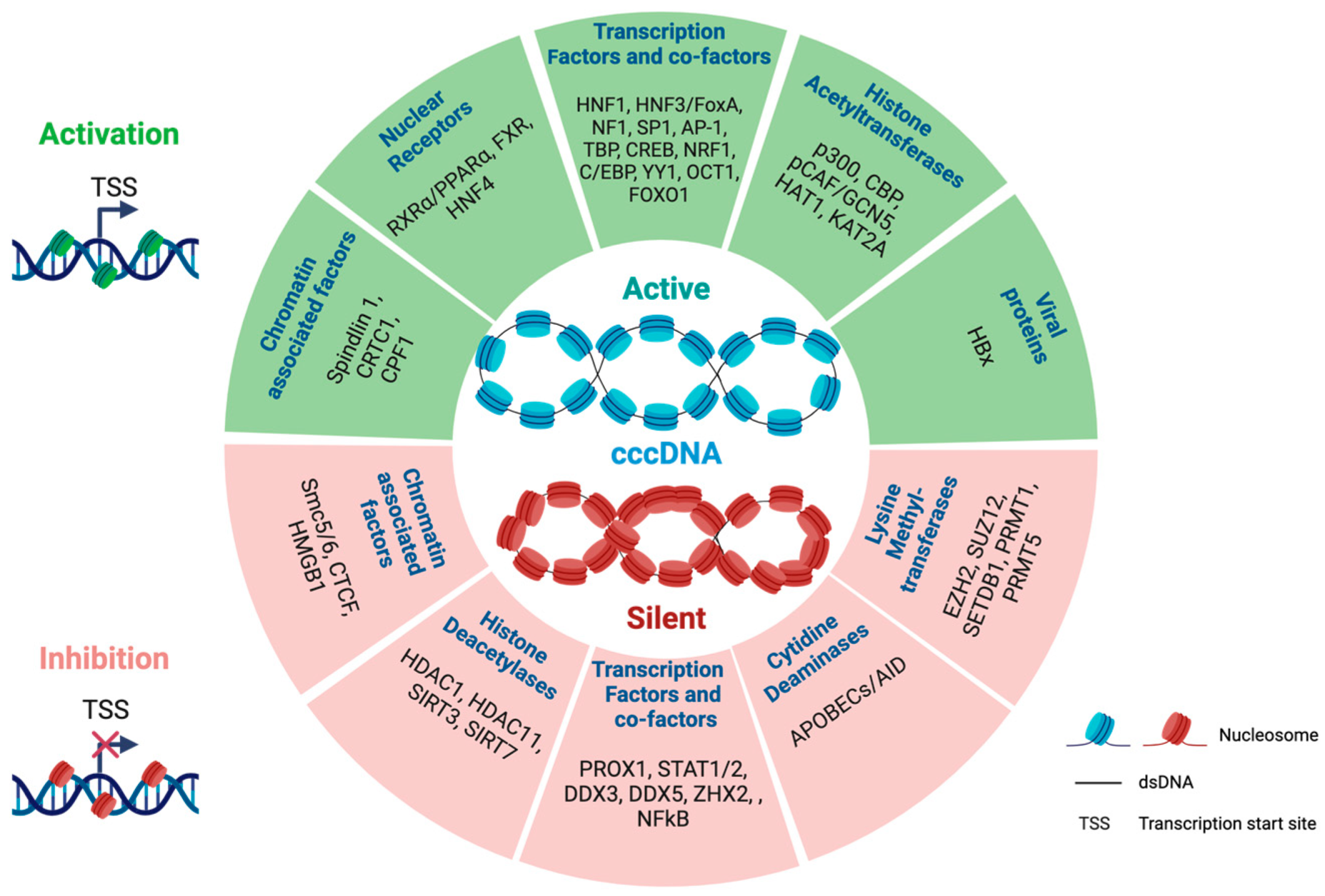
2.3. Trans-Factors Controlling cccDNA Transcriptional Activity
2.4. Epigenetic Modulation of cccDNA Transcriptional Activity
2.5. Compartmentalization of cccDNA in the Nucleus of Infected Hepatocytes

3. Co-Transcriptional Regulation of HBV RNAs
3.1. HBV RNA Splicing
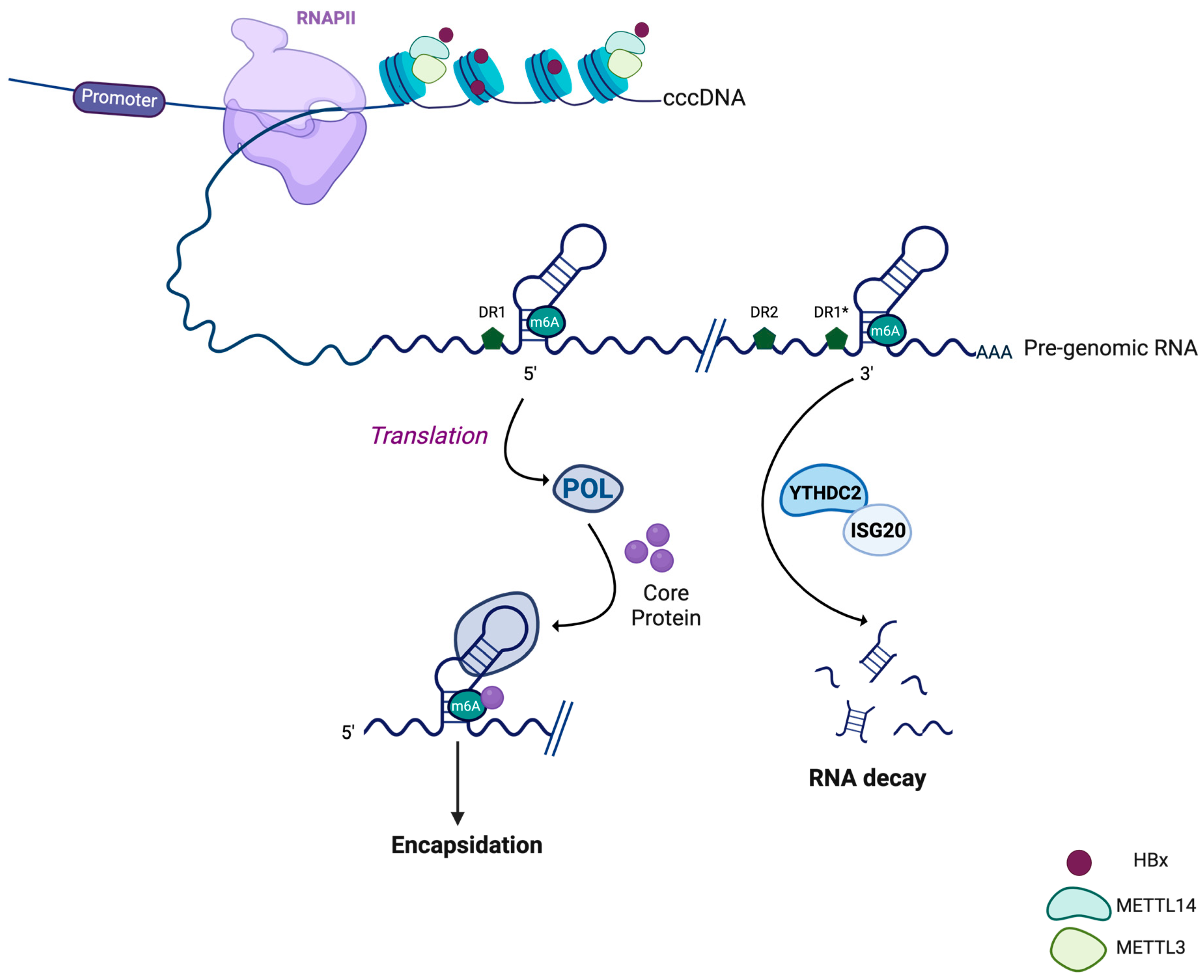
3.2. HBV RNA Methylation
3.3. HBV RNA Polyadenylation
3.4. Capping of HBV RNAs
4. Conclusions
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the Management of Hepatitis B Virus Infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Boyd, A.; Combe, E.; Testoni, B.; Zoulim, F. Covalently Closed Circular DNA: The Ultimate Therapeutic Target for Curing HBV Infections. J. Hepatol. 2021, 75, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Testoni, B.; Zoulim, F. Biological Basis for Functional Cure of Chronic Hepatitis B. J. Viral Hepat. 2019, 26, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Combe, E.; Inchauspe, A.; Mangeot, P.E.; Delberghe, E.; Chapus, F.; Neveu, G.; Alam, A.; Carter, K.; Testoni, B.; et al. CRISPR-Cas9 Targeting of Hepatitis B Virus Covalently Closed Circular DNA Generates Transcriptionally Active Episomal Variants. mBio 2022, 13, e0288821. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Long, K.R.; Loprieno, M.A.; De Silva Feelixge, H.S.; Kenkel, E.J.; Liley, R.M.; Rapp, S.; Roychoudhury, P.; Nguyen, T.; Stensland, L.; et al. CRISPR-Cas9 Gene Editing of Hepatitis B Virus in Chronically Infected Humanized Mice. Mol. Ther. Methods Clin. Dev. 2021, 20, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Gorsuch, C.L.; Nemec, P.; Yu, M.; Xu, S.; Han, D.; Smith, J.; Lape, J.; van Buuren, N.; Ramirez, R.; Muench, R.C.; et al. Targeting the Hepatitis B cccDNA with a Sequence-Specific ARCUS Nuclease to Eliminate Hepatitis B Virus in Vivo. Mol. Ther. 2022, 30, 2909–2922. [Google Scholar] [CrossRef] [PubMed]
- Smekalova, E.M.; Martinez, M.G.; Combe, E.; Kumar, A.; Dejene, S.; Leboeuf, D.; Chen, C.-Y.; Dorkin, J.R.; Shuang, L.S.; Kieft, S.; et al. Cytosine Base Editing Inhibits Hepatitis B Virus Replication and Reduces HBsAg Expression in vitro and in vivo. Mol. Ther. Nucleic Acids 2024, 35, 102112. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Smekalova, E.; Combe, E.; Gregoire, F.; Zoulim, F.; Testoni, B. Gene Editing Technologies to Target HBV cccDNA. Viruses 2022, 14, 2654. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Chen, J. Interferon and Hepatitis B: Current and Future Perspectives. Front. Immunol. 2021, 12, 733364. [Google Scholar] [CrossRef]
- Mohd-Ismail, N.K.; Lim, Z.; Gunaratne, J.; Tan, Y.-J. Mapping the Interactions of HBV cccDNA with Host Factors. Int. J. Mol. Sci. 2019, 20, 4276. [Google Scholar] [CrossRef]
- Wei, L.; Ploss, A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses 2021, 13, 1463. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.T.; Schranz, P.; Schröder, C.H.; Zentgraf, H. Hepatitis B Virus Genome Is Organized into Nucleosomes in the Nucleus of the Infected Cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Stadelmayer, B.; Diederichs, A.; Chapus, F.; Rivoire, M.; Neveu, G.; Alam, A.; Fraisse, L.; Carter, K.; Testoni, B.; Zoulim, F. Full-Length 5′RACE Identifies All Major HBV Transcripts in HBV-Infected Hepatocytes and Patient Serum. J. Hepatol. 2020, 73, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Altinel, K.; Hashimoto, K.; Wei, Y.; Neuveut, C.; Gupta, I.; Suzuki, A.M.; Dos Santos, A.; Moreau, P.; Xia, T.; Kojima, S.; et al. Single-Nucleotide Resolution Mapping of Hepatitis B Virus Promoters in Infected Human Livers and Hepatocellular Carcinoma. J. Virol. 2016, 90, 10811–10822. [Google Scholar] [CrossRef] [PubMed]
- Quarleri, J. Core Promoter: A Critical Region Where the Hepatitis B Virus Makes Decisions. World J. Gastroenterol. 2014, 20, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A.; Kew, M.C. The Core Promoter of Hepatitis B Virus. J. Viral Hepat. 1999, 6, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Mertz, J.E. Promoters for Synthesis of the Pre-C and Pregenomic MRNAs of Human Hepatitis B Virus Are Genetically Distinct and Differentially Regulated. J. Virol. 1996, 70, 8719–8726. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, R.; Will, H.; Hernandez, N.; Schaller, H. Signals Regulating Hepatitis B Surface Antigen Transcription. Nature 1983, 305, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.K.; Ting, L.P. The Surface Gene Promoter of the Human Hepatitis B Virus Displays a Preference for Differentiated Hepatocytes. Virology 1989, 170, 176–183. [Google Scholar] [CrossRef]
- Pourcel, C.; Louise, A.; Gervais, M.; Chenciner, N.; Dubois, M.F.; Tiollais, P. Transcription of the Hepatitis B Surface Antigen Gene in Mouse Cells Transformed with Cloned Viral DNA. J. Virol. 1982, 42, 100–105. [Google Scholar] [CrossRef]
- De-Medina, T.; Faktor, O.; Shaul, Y. The S Promoter of Hepatitis B Virus Is Regulated by Positive and Negative Elements. Mol. Cell. Biol. 1988, 8, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Koike, K. Identification of a Binding Protein to the X Gene Promoter Region of Hepatitis B Virus. Virology 1992, 191, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, K.; Nakamura, I.; Takada, S.; Koike, K. A Transcription Initiation Site for the Hepatitis B Virus X Gene Is Directed by the Promoter-Binding Protein. J. Virol. 1993, 67, 2559–2565. [Google Scholar] [CrossRef] [PubMed]
- Treinin, M.; Laub, O. Identification of a Promoter Element Located Upstream from the Hepatitis B Virus X Gene. Mol. Cell. Biol. 1987, 7, 545–548. [Google Scholar] [CrossRef]
- Chang, H.K.; Chou, C.K.; Chang, C.; Su, T.S.; Hu, C.; Yoshida, M.; Ting, L.P. The Enhancer Sequence of Human Hepatitis B Virus Can Enhance the Activity of Its Surface Gene Promoter. Nucleic Acids Res. 1987, 15, 2261–2268. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, K.Q.; Siddiqui, A. Regulation of the Hepatitis B Virus Gene Expression by the Enhancer Element I. Virology 1991, 181, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.; Rutter, W.J.; Laub, O. A Human Hepatitis B Viral Enhancer Element. EMBO J. 1985, 4, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Yee, J.K. A Liver-Specific Enhancer in the Core Promoter Region of Human Hepatitis B Virus. Science 1989, 246, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Yuh, C.H.; Ting, L.P. The Genome of Hepatitis B Virus Contains a Second Enhancer: Cooperation of Two Elements within This Enhancer Is Required for Its Function. J. Virol. 1990, 64, 4281–4287. [Google Scholar] [CrossRef]
- Doitsh, G.; Shaul, Y. Enhancer I Predominance in Hepatitis B Virus Gene Expression. Mol. Cell. Biol. 2004, 24, 1799–1808. [Google Scholar] [CrossRef]
- Biswas, B.; Kandpal, M.; Vivekanandan, P. A G-Quadruplex Motif in an Envelope Gene Promoter Regulates Transcription and Virion Secretion in HBV Genotype B. Nucleic Acids Res. 2017, 45, 11268–11280. [Google Scholar] [CrossRef] [PubMed]
- Saranathan, N.; Vivekanandan, P. G-Quadruplexes: More Than Just a Kink in Microbial Genomes. Trends Microbiol. 2019, 27, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Meier-Stephenson, V.; Badmalia, M.D.; Mrozowich, T.; Lau, K.C.K.; Schultz, S.K.; Gemmill, D.L.; Osiowy, C.; van Marle, G.; Coffin, C.S.; Patel, T.R. Identification and Characterization of a G-Quadruplex Structure in the Pre-Core Promoter Region of Hepatitis B Virus Covalently Closed Circular DNA. J. Biol. Chem. 2021, 296, 100589. [Google Scholar] [CrossRef] [PubMed]
- Giraud, G.; Rodà, M.; Huchon, P.; Michelet, M.; Maadadi, S.; Jutzi, D.; Montserret, R.; Ruepp, M.-D.; Parent, R.; Combet, C.; et al. G-Quadruplexes Control Hepatitis B Virus Replication by Promoting cccDNA Transcription and Phase Separation in Hepatocytes. Nucleic Acids Res. 2024, 52, 2290–2305. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B Virus X Protein Is Essential to Initiate and Maintain Virus Replication after Infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Bouchard, M.J. Role of HBx in Hepatitis B Virus Persistence and Its Therapeutic Implications. Curr. Opin. Virol. 2018, 30, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Decorsière, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B Virus X Protein Identifies the Smc5/6 Complex as a Host Restriction Factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef]
- Livingston, C.M.; Ramakrishnan, D.; Strubin, M.; Fletcher, S.P.; Beran, R.K. Identifying and Characterizing Interplay between Hepatitis B Virus X Protein and Smc5/6. Viruses 2017, 9, 69. [Google Scholar] [CrossRef]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular Functions and Biological Roles of Hepatitis B Virus x Protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef]
- Chong, C.K.; Cheng, C.Y.S.; Tsoi, S.Y.J.; Huang, F.-Y.; Liu, F.; Seto, W.-K.; Lai, C.-L.; Yuen, M.-F.; Wong, D.K.-H. Role of Hepatitis B Core Protein in HBV Transcription and Recruitment of Histone Acetyltransferases to cccDNA Minichromosome. Antivir. Res. 2017, 144, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Zehnder, B.; Qu, B.; Urban, S. De Novo Synthesis of Hepatitis B Virus Nucleocapsids Is Dispensable for the Maintenance and Transcriptional Regulation of cccDNA. JHEP Rep. 2021, 3, 100195. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kang, H.S.; Kim, K.-H. Roles of Hepatocyte Nuclear Factors in Hepatitis B Virus Infection. World J. Gastroenterol. 2016, 22, 7017–7029. [Google Scholar] [CrossRef]
- Teng, Y.; Xu, Z.; Zhao, K.; Zhong, Y.; Wang, J.; Zhao, L.; Zheng, Z.; Hou, W.; Zhu, C.; Chen, X.; et al. Novel Function of SART1 in HNF4α Transcriptional Regulation Contributes to Its Antiviral Role during HBV Infection. J. Hepatol. 2021, 75, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhou, W.; Zhu, H.; Zhou, P.; Shi, X. Baicalin Benefits the Anti-HBV Therapy via Inhibiting HBV Viral RNAs. Toxicol. Appl. Pharmacol. 2017, 323, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Shlomai, A.; Paran, N.; Shaul, Y. PGC-1α Controls Hepatitis B Virus through Nutritional Signals. Proc. Natl. Acad. Sci. USA 2006, 103, 16003–16008. [Google Scholar] [CrossRef] [PubMed]
- Shlomai, A.; Shaul, Y. The Metabolic Activator FOXO1 Binds Hepatitis B Virus DNA and Activates Its Transcription. Biochem. Biophys. Res. Commun. 2009, 381, 544–548. [Google Scholar] [CrossRef]
- D’Arienzo, V.; Ferguson, J.; Giraud, G.; Chapus, F.; Harris, J.M.; Wing, P.A.C.; Claydon, A.; Begum, S.; Zhuang, X.; Balfe, P.; et al. The CCCTC-Binding Factor CTCF Represses Hepatitis B Virus Enhancer I and Regulates Viral Transcription. Cell. Microbiol. 2021, 23, e13274. [Google Scholar] [CrossRef]
- Dobrica, M.O.; Varghese, C.S.; Harris, J.M.; Ferguson, J.; Magri, A.; Arnold, R.; Várnai, C.; Parish, J.L.; McKeating, J.A. CTCF Regulates Hepatitis B Virus cccDNA Chromatin Topology. J. Gen. Virol. 2024, 105, 001939. [Google Scholar] [CrossRef]
- Ren, F.; Ren, J.-H.; Song, C.-L.; Tan, M.; Yu, H.-B.; Zhou, Y.-J.; Qin, Y.-P.; Cheng, S.-T.; Zhang, Y.; Huang, A.-L.; et al. LncRNA HOTAIR Modulates Hepatitis B Virus Transcription and Replication by Enhancing SP1 Transcription Factor. Clin. Sci. 2020, 134, 3007–3022. [Google Scholar] [CrossRef]
- Shaul, Y.; Ben-Levy, R. Multiple Nuclear Proteins in Liver Cells Are Bound to Hepatitis B Virus Enhancer Element and Its Upstream Sequences. EMBO J. 1987, 6, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Turton, K.L.; Meier-Stephenson, V.; Badmalia, M.D.; Coffin, C.S.; Patel, T.R. Host Transcription Factors in Hepatitis B Virus RNA Synthesis. Viruses 2020, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, J.; Wu, M.; Zhang, X.; Zhang, M.; Yue, L.; Li, Y.; Liu, J.; Li, B.; Shen, F.; et al. PRMT5 Restricts Hepatitis B Virus Replication through Epigenetic Repression of Covalently Closed Circular DNA Transcription and Interference with Pregenomic RNA Encapsidation. Hepatology 2017, 66, 398–415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xing, Z.; Mani, S.K.K.; Bancel, B.; Durantel, D.; Zoulim, F.; Tran, E.J.; Merle, P.; Andrisani, O. RNA Helicase DEAD Box Protein 5 Regulates Polycomb Repressive Complex 2/Hox Transcript Antisense Intergenic RNA Function in Hepatitis B Virus Infection and Hepatocarcinogenesis. Hepatology 2016, 64, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Feng, X.; Mao, T.; Yang, D.; Zou, J.; Zao, X.; Deng, Q.; Chen, X.; Lu, F. Yin-Yang 1 and HBx Protein Activate HBV Transcription by Mediating the Spatial Interaction of cccDNA Minichromosome with Cellular Chromosome 19p13.11. Emerg. Microbes Infect. 2020, 9, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhao, K.; Yao, Y.; Liu, C.; Chen, Y.; Li, J.; Wang, Y.; Pei, R.; Chen, J.; Hu, X.; et al. HDAC11 Restricts HBV Replication through Epigenetic Repression of cccDNA Transcription. Antivir. Res. 2019, 172, 104619. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Feng, J.; Liu, Y.; Zhao, M.; Yuan, Y.; Yuan, H.; Yun, H.; Sun, M.; Bu, Y.; Liu, L.; et al. HAT1 Signaling Confers to Assembly and Epigenetic Regulation of HBV cccDNA Minichromosome. Theranostics 2019, 9, 7345–7358. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-B.; Cheng, S.-T.; Ren, F.; Chen, Y.; Shi, X.-F.; Wong, V.K.W.; Law, B.Y.K.; Ren, J.-H.; Zhong, S.; Chen, W.-X.; et al. SIRT7 Restricts HBV Transcription and Replication through Catalyzing Desuccinylation of Histone H3 Associated with cccDNA Minichromosome. Clin. Sci. 2021, 135, 1505–1522. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.-P.; Yu, H.-B.; Yuan, S.-Y.; Yang, Z.; Ren, F.; Wang, Q.; Li, F.; Ren, J.-H.; Cheng, S.-T.; Zhou, Y.-J.; et al. KAT2A Promotes Hepatitis B Virus Transcription and Replication Through Epigenetic Regulation of cccDNA Minichromosome. Front. Microbiol. 2022, 12, 795388. [Google Scholar] [CrossRef]
- Rivière, L.; Gerossier, L.; Ducroux, A.; Dion, S.; Deng, Q.; Michel, M.-L.; Buendia, M.-A.; Hantz, O.; Neuveut, C. HBx Relieves Chromatin-Mediated Transcriptional Repression of Hepatitis B Viral cccDNA Involving SETDB1 Histone Methyltransferase. J. Hepatol. 2015, 63, 1093–1102. [Google Scholar] [CrossRef]
- Salerno, D.; Chiodo, L.; Alfano, V.; Floriot, O.; Cottone, G.; Paturel, A.; Pallocca, M.; Plissonnier, M.-L.; Jeddari, S.; Belloni, L.; et al. Hepatitis B Protein HBx Binds the DLEU2 LncRNA to Sustain cccDNA and Host Cancer-Related Gene Transcription. Gut 2020, 69, 2016–2024. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Teng, Y.; Wang, L.; Zhang, Z.; Chen, C.; Wang, Y.; Zhang, X.; Xiang, P.; Song, X.; Lu, J.; et al. LINC01431 Promotes Histone H4R3 Methylation to Impede HBV Covalently Closed Circular DNA Transcription by Stabilizing PRMT1. Adv. Sci. 2022, 9, e2103135. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx Binds the HBV Minichromosome and Modifies the Epigenetic Regulation of cccDNA Function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.K.; Cheng, C.Y.S.; Tsoi, S.Y.J.; Huang, F.-Y.; Liu, F.; Fung, J.; Seto, W.-K.; Lai, K.K.-Y.; Lai, C.-L.; Yuen, M.-F.; et al. HBV X Protein Mutations Affect HBV Transcription and Association of Histone-Modifying Enzymes with Covalently Closed Circular DNA. Sci. Rep. 2020, 10, 802. [Google Scholar] [CrossRef] [PubMed]
- Cougot, D.; Wu, Y.; Cairo, S.; Caramel, J.; Renard, C.-A.; Lévy, L.; Buendia, M.A.; Neuveut, C. The Hepatitis B Virus X Protein Functionally Interacts with CREB-Binding Protein/P300 in the Regulation of CREB-Mediated Transcription. J. Biol. Chem. 2007, 282, 4277–4287. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yao, Q.; Su, X.; Deng, Y.; Yang, M.; Peng, B.; Zhao, F.; Du, C.; Zhang, X.; Zhu, J.; et al. Molecular Insights into Spindlin1-HBx Interplay and Its Impact on HBV Transcription from cccDNA Minichromosome. Nat. Commun. 2023, 14, 4663. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Zhou, J.; Zhang, H.; Marchetti, A.; van de Klundert, M.; Cai, D.; Yu, X.; Mitra, B.; Liu, Y.; Wang, M.; et al. Hepatitis B Virus X Protein Counteracts High Mobility Group Box 1 Protein-Mediated Epigenetic Silencing of Covalently Closed Circular DNA. PLoS Pathog. 2022, 18, e1010576. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Yuan, H.; Yang, G.; Yun, H.; Zhao, M.; Liu, Z.; Zhao, L.; Geng, Y.; Liu, L.; Wang, J.; et al. IFN-α Confers Epigenetic Regulation of HBV cccDNA Minichromosome by Modulating GCN5-Mediated Succinylation of Histone H3K79 to Clear HBV cccDNA. Clin. Epigenetics 2020, 12, 135. [Google Scholar] [CrossRef] [PubMed]
- Lebossé, F.; Inchauspé, A.; Locatelli, M.; Miaglia, C.; Diederichs, A.; Fresquet, J.; Chapus, F.; Hamed, K.; Testoni, B.; Zoulim, F. Quantification and Epigenetic Evaluation of the Residual Pool of Hepatitis B Covalently Closed Circular DNA in Long-Term Nucleoside Analogue-Treated Patients. Sci. Rep. 2020, 10, 21097. [Google Scholar] [CrossRef]
- Moreau, P.; Cournac, A.; Palumbo, G.A.; Marbouty, M.; Mortaza, S.; Thierry, A.; Cairo, S.; Lavigne, M.; Koszul, R.; Neuveut, C. Tridimensional Infiltration of DNA Viruses into the Host Genome Shows Preferential Contact with Active Chromatin. Nat. Commun. 2018, 9, 4268. [Google Scholar] [CrossRef]
- Tang, D.; Zhao, H.; Wu, Y.; Peng, B.; Gao, Z.; Sun, Y.; Duan, J.; Qi, Y.; Li, Y.; Zhou, Z.; et al. Transcriptionally Inactive Hepatitis B Virus Episome DNA Preferentially Resides in the Vicinity of Chromosome 19 in 3D Host Genome upon Infection. Cell Rep. 2021, 35, 109288. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.D.; Sarica, N.; Cournac, A.; Koszul, R.; Neuveut, C. Crosstalk between Hepatitis B Virus and the 3D Genome Structure. Viruses 2022, 14, 445. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, E.-M.; Xu, L.-Y. From Start to End: Phase Separation and Transcriptional Regulation. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2020, 1863, 194641. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Lee, S.; Windisch, M.P.; Ryu, W.S. DDX3 Dead-box RNA helicase is a host factor that restricts hepatitis B virus replication at the transcriptional level. J. Virol. 2014, 88, 13689–13698. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.V.; Gao, W.W.; Chan, C.P.; Cheng, Y.; Chaudhary, V.; Deng, J.J.; Yuen, K.S.; Wong, C.M.; Ng, I.O.; Kok, K.H.; et al. Requirement of CRTC1 coactivator for hepatitis B virus transcription. Nucleic Acids Res. 2014, 42, 12455–12468. [Google Scholar] [CrossRef]
- Ren, J.H.; Hu, J.L.; Cheng, S.T.; Yu, H.B.; Wong, V.K.W.; Law, B.Y.K.; Yang, Y.F.; Huang, Y.; Liu, Y.; Chen, W.X.; et al. SIRT3 restricts hepatitis B virus transcription and replication through epigenetic regulation of covalently closed circular DNA involving suppressor of variegation 3-9 homolog 1 and SET domain containing 1A histone methyltransferases. Hepatology 2018, 68, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Kostyushev, D.; Brezgin, S.; Kostyusheva, A.; Ponomareva, N.; Bayurova, E.; Zakirova, N.; Kondrashova, A.; Goptar, I.; Nikiforova, A.; Sudina, A.; et al. Transient and tunable CRISPRa regulation of APOBEC/AID genes for targeting hepatitis B virus. Mol. Ther. Nucleic Acids. 2023, 20, 478–493. [Google Scholar] [CrossRef]
- Kremsdorf, D.; Lekbaby, B.; Bablon, P.; Sotty, J.; Augustin, J.; Schnuriger, A.; Pol, J.; Soussan, P. Alternative Splicing of Viral Transcripts: The Dark Side of HBV. Gut 2021, 70, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, M.; Wang, F.; Zhang, W.; Wang, W.; Zhang, X.; Zhang, J.; Liu, Y.; Liu, Y.; Feng, Y.; et al. Hepatitis B Virus Spliced Variants Are Associated with an Impaired Response to Interferon Therapy. Sci. Rep. 2015, 5, 16459. [Google Scholar] [CrossRef]
- Candotti, D.; Allain, J.-P. Biological and Clinical Significance of Hepatitis B Virus RNA Splicing: An Update. Ann. Blood 2017, 2, 6. [Google Scholar] [CrossRef]
- Francies, F.Z.; Dlamini, Z. Aberrant Splicing Events and Epigenetics in Viral Oncogenomics: Current Therapeutic Strategies. Cells 2021, 10, 239. [Google Scholar] [CrossRef] [PubMed]
- Su, T.S.; Lai, C.J.; Huang, J.L.; Lin, L.H.; Yauk, Y.K.; Chang, C.M.; Lo, S.J.; Han, S.H. Hepatitis B Virus Transcript Produced by RNA Splicing. J. Virol. 1989, 63, 4011–4018. [Google Scholar] [CrossRef] [PubMed]
- Su, T.-S.; Lui, W.-Y.; Lin, L.-H.; Han, S.-H.; P’eng, F.-K. Analysis of Hepatitis B Virus Transcripts in Infected Human Livers. Hepatology 1989, 9, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Hass, M.; Hannoun, C.; Kalinina, T.; Sommer, G.; Manegold, C.; Günther, S. Functional Analysis of Hepatitis B Virus Reactivating in Hepatitis B Surface Antigen-Negative Individuals. Hepatology 2005, 42, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Köck, J.; Blum, H.E. A Novel Target of Hepatitis B Virus Mutations: Splicing of Surface RNA. Hepatology 2005, 42, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Heise, T.; Sommer, G.; Reumann, K.; Meyer, I.; Will, H.; Schaal, H. The Hepatitis B Virus PRE Contains a Splicing Regulatory Element. Nucleic Acids Res. 2006, 34, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Xie, M.-H.; Liu, W.; Yang, B.; Yang, F.; Huang, J.; Huang, J.; Wu, Q.; Fu, X.-D.; Zhang, Y. A Structured RNA in Hepatitis B Virus Post-Transcriptional Regulatory Element Represses Alternative Splicing in a Sequence-Independent and Position-Dependent Manner. FEBS J. 2011, 278, 1533–1546. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Liao, G.; Zhang, M.; Zhu, Y.; Wang, K.; Xiao, W.; Jia, C.; Dong, M.; Sun, N.; Walch, A.; et al. Translatomic Profiling Reveals Novel Self-Restricting Virus-Host Interactions during HBV Infection. J. Hepatol. 2021, 75, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Taha, T.Y.; Anirudhan, V.; Limothai, U.; Loeb, D.D.; Petukhov, P.A.; McLachlan, A. Modulation of Hepatitis B Virus Pregenomic RNA Stability and Splicing by Histone Deacetylase 5 Enhances Viral Biosynthesis. PLOS Pathog. 2020, 16, e1008802. [Google Scholar] [CrossRef]
- Soussan, P.; Pol, J.; Garreau, F.; Schneider, V.; Le Pendeven, C.; Nalpas, B.; Lacombe, K.; Bonnard, P.; Pol, S.; Kremsdorf, D. Expression of Defective Hepatitis B Virus Particles Derived from Singly Spliced RNA Is Related to Liver Disease. J. Infect. Dis. 2008, 198, 218–225. [Google Scholar] [CrossRef]
- Bayliss, J.; Lim, L.; Thompson, A.J.V.; Desmond, P.; Angus, P.; Locarnini, S.; Revill, P.A. Hepatitis B Virus Splicing Is Enhanced Prior to Development of Hepatocellular Carcinoma. J. Hepatol. 2013, 59, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, M.; Zhang, X.; Zhang, W.; Zhang, Z.; Chen, L.; He, J.; Zheng, Y.; Chen, C.; Wang, F.; et al. Hepatitis B Virus Polymerase Impairs Interferon-α-Induced STA T Activation through Inhibition of Importin-A5 and Protein Kinase C-δ. Hepatology 2013, 57, 470–482. [Google Scholar] [CrossRef]
- Yu, S.; Chen, J.; Wu, M.; Chen, H.; Kato, N.; Yuan, Z. Hepatitis B Virus Polymerase Inhibits RIG-I- and Toll-like Receptor 3-Mediated Beta Interferon Induction in Human Hepatocytes through Interference with Interferon Regulatory Factor 3 Activation and Dampening of the Interaction between TBK1/IKKepsilon and DDX3. J. Gen. Virol. 2010, 91, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Mancini-Bourgine, M.; Bayard, F.; Soussan, P.; Deng, Q.; Lone, Y.-C.; Kremsdorf, D.; Michel, M.-L. Hepatitis B Virus Splice-Generated Protein Induces T-Cell Responses in HLA-Transgenic Mice and Hepatitis B Virus-Infected Patients. J. Virol. 2007, 81, 4963–4972. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Y.; Chen, W.; Zheng, X.; Guo, Y.; Cao, J.; Zhang, Y.; Wen, S.; Gao, W.; Wu, Y. Regulatory Role and Mechanism of M6A RNA Modification in Human Metabolic Diseases. Mol. Ther.—Oncolytics 2021, 22, 52–63. [Google Scholar] [CrossRef]
- Tirumuru, N.; Zhao, B.S.; Lu, W.; Lu, Z.; He, C.; Wu, L. N6-Methyladenosine of HIV-1 RNA Regulates Viral Infection and HIV-1 Gag Protein Expression. eLife 2016, 5, e15528. [Google Scholar] [CrossRef] [PubMed]
- Imam, H.; Khan, M.; Gokhale, N.S.; McIntyre, A.B.R.; Kim, G.-W.; Jang, J.Y.; Kim, S.-J.; Mason, C.E.; Horner, S.M.; Siddiqui, A. N6-Methyladenosine Modification of Hepatitis B Virus RNA Differentially Regulates the Viral Life Cycle. Proc. Natl. Acad. Sci. USA 2018, 115, 8829–8834. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-W.; Siddiqui, A. Hepatitis B Virus X Protein Recruits Methyltransferases to Affect Cotranscriptional N6-Methyladenosine Modification of Viral/Host RNAs. Proc. Natl. Acad. Sci. USA 2021, 118, e2019455118. [Google Scholar] [CrossRef]
- Chakraborty, D.; Ghosh, S. The Epsilon Motif of Hepatitis B Virus RNA Exhibits a Potassium-Dependent Ribonucleolytic Activity. FEBS J. 2017, 284, 1184–1203. [Google Scholar] [CrossRef]
- Fleming, A.M.; Nguyen, N.L.B.; Burrows, C.J. Colocalization of M6A and G-Quadruplex-Forming Sequences in Viral RNA (HIV, Zika, Hepatitis B, and SV40) Suggests Topological Control of Adenosine N6-Methylation. ACS Cent. Sci. 2019, 5, 218–228. [Google Scholar] [CrossRef]
- Yoshida, A.; Oyoshi, T.; Suda, A.; Futaki, S.; Imanishi, M. Recognition of G-Quadruplex RNA by a Crucial RNA Methyltransferase Component, METTL14. Nucleic Acids Res. 2022, 50, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-W.; Moon, J.-S.; Siddiqui, A. N6-Methyladenosine Modification of the 5′ Epsilon Structure of the HBV Pregenome RNA Regulates Its Encapsidation by the Viral Core Protein. Proc. Natl. Acad. Sci. USA 2022, 119, e2120485119. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-W.; Imam, H.; Siddiqui, A. The RNA Binding Proteins YTHDC1 and FMRP Regulate the Nuclear Export of N6-Methyladenosine-Modified Hepatitis B Virus Transcripts and Affect the Viral Life Cycle. J. Virol. 2021, 95, e0009721. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-W.; Imam, H.; Khan, M.; Siddiqui, A. N6-Methyladenosine Modification of Hepatitis B and C Viral RNAs Attenuates Host Innate Immunity via RIG-I Signaling. J. Biol. Chem. 2020, 295, 13123–13133. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-W.; Siddiqui, A. Hepatitis B Virus X Protein Expression Is Tightly Regulated by N6-Methyladenosine Modification of Its MRNA. J. Virol. 2022, 96, e0165521. [Google Scholar] [CrossRef] [PubMed]
- Baginski, I.; Chemin, I.; Bouffard, P.; Hantz, O.; Trepo, C. Detection of Polyadenylated RNA in Hepatitis B Virus-Infected Peripheral Blood Mononuclear Cells by Polymerase Chain Reaction. J. Infect. Dis. 1991, 163, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, C.C.; Levinson, A.D. Analysis of Processing and Polyadenylation Signals of the Hepatitis B Virus Surface Antigen Gene by Using Simian Virus 40-Hepatitis B Virus Chimeric Plasmids. Mol. Cell. Biol. 1983, 3, 2250–2258. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Shaul, Y. A Long HBV Transcript Encoding PX Is Inefficiently Exported from the Nucleus. Virology 2003, 309, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Russnak, R.H. Regulation of Polyadenylation in Hepatitis B Viruses: Stimulation by the Upstream Activating Signal PS1 Is Orientation-Dependent, Distance-Independent, and Additive. Nucleic Acids Res. 1991, 19, 6449–6456. [Google Scholar] [CrossRef]
- Russnak, R.; Ganem, D. Sequences 5′ to the Polyadenylation Signal Mediate Differential Poly(A) Site Use in Hepatitis B Viruses. Genes Dev. 1990, 4, 764–776. [Google Scholar] [CrossRef]
- Chapus, F.; Giraud, G.; Huchon, P.; Charre, C.; Goldsmith, C.; Rodà, M.; Martinez, M.G.; Fresquet, J.; Diederichs, A.; Locatelli, M.; et al. Helicases DDX5 and DDX17 Promote Hepatitis B Virus Transcription Termination Heterogeneity in Infected Human Hepatocytes. bioRxiv 2024. [Google Scholar] [CrossRef]
- Paran, N.; Ori, A.; Haviv, I.; Shaul, Y. A Composite Polyadenylation Signal with TATA Box Function. Mol. Cell. Biol. 2000, 20, 834–841. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kang, W.; Ha, K.S.; Uhm, H.; Park, K.; Lee, J.Y.; Hohng, S.; Kang, C. Transcription Reinitiation by Recycling RNA Polymerase That Diffuses on DNA after Releasing Terminated RNA. Nat. Commun. 2020, 11, 450. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Mapendano, C.K.; Jensen, T.H. An Ending Is a New Beginning: Transcription Termination Supports Re-Initiation. Cell Cycle 2011, 10, 863–865. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cavallaro, M.; Walsh, M.D.; Jones, M.; Teahan, J.; Tiberi, S.; Finkenstädt, B.; Hebenstreit, D. 3′-5′ Crosstalk Contributes to Transcriptional Bursting. Genome Biol. 2021, 22, 56. [Google Scholar] [CrossRef] [PubMed]
- Hyrina, A.; Jones, C.; Chen, D.; Clarkson, S.; Cochran, N.; Feucht, P.; Hoffman, G.; Lindeman, A.; Russ, C.; Sigoillot, F.; et al. A Genome-Wide CRISPR Screen Identifies ZCCHC14 as a Host Factor Required for Hepatitis B Surface Antigen Production. Cell Rep. 2019, 29, 2970–2978.e6. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lee, A.C.H.; Guo, F.; Kondratowicz, A.S.; Micolochick Steuer, H.M.; Miller, A.; Bailey, L.D.; Wang, X.; Chen, S.; Kultgen, S.G.; et al. Host Poly(A) Polymerases PAPD5 and PAPD7 Provide Two Layers of Protection That Ensure the Integrity and Stability of Hepatitis B Virus RNA. J. Virol. 2021, 95, e0057421. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A.; Robb, G.B.; Chan, S.-H. MRNA Capping: Biological Functions and Applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef] [PubMed]
- Galloway, A.; Cowling, V.H. MRNA Cap Regulation in Mammalian Cell Function and Fate. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2019, 1862, 270–279. [Google Scholar] [CrossRef]
- Junker-Niepmann, M.; Bartenschlager, R.; Schaller, H. A Short Cis-Acting Sequence Is Required for Hepatitis B Virus Pregenome Encapsidation and Sufficient for Packaging of Foreign RNA. EMBO J. 1990, 9, 3389–3396. [Google Scholar] [CrossRef]
- Jeong, J.K.; Yoon, G.S.; Ryu, W.S. Evidence That the 5′-End Cap Structure Is Essential for Encapsidation of Hepatitis B Virus Pregenomic RNA. J. Virol. 2000, 74, 5502–5508. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Wang, H.; Ryu, W.-S. Incorporation of Eukaryotic Translation Initiation Factor EIF4E into Viral Nucleocapsids via Interaction with Hepatitis B Virus Polymerase. J. Virol. 2010, 84, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-Molecule Inhibition of METTL3 as a Strategy against Myeloid Leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Mueller, H.; Wildum, S.; Luangsay, S.; Walther, J.; Lopez, A.; Tropberger, P.; Ottaviani, G.; Lu, W.; Parrott, N.J.; Zhang, J.D.; et al. A Novel Orally Available Small Molecule That Inhibits Hepatitis B Virus Expression. J. Hepatol. 2018, 68, 412–420. [Google Scholar] [CrossRef]
- Menne, S.; Wildum, S.; Steiner, G.; Suresh, M.; Korolowicz, K.; Balarezo, M.; Yon, C.; Murreddu, M.; Hong, X.; Kallakury, B.V.; et al. Efficacy of an Inhibitor of Hepatitis B Virus Expression in Combination With Entecavir and Interferon-α in Woodchucks Chronically Infected With Woodchuck Hepatitis Virus. Hepatol. Commun. 2020, 4, 916–931. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giraud, G.; El Achi, K.; Zoulim, F.; Testoni, B. Co-Transcriptional Regulation of HBV Replication: RNA Quality Also Matters. Viruses 2024, 16, 615. https://doi.org/10.3390/v16040615
Giraud G, El Achi K, Zoulim F, Testoni B. Co-Transcriptional Regulation of HBV Replication: RNA Quality Also Matters. Viruses. 2024; 16(4):615. https://doi.org/10.3390/v16040615
Chicago/Turabian StyleGiraud, Guillaume, Khadija El Achi, Fabien Zoulim, and Barbara Testoni. 2024. "Co-Transcriptional Regulation of HBV Replication: RNA Quality Also Matters" Viruses 16, no. 4: 615. https://doi.org/10.3390/v16040615
APA StyleGiraud, G., El Achi, K., Zoulim, F., & Testoni, B. (2024). Co-Transcriptional Regulation of HBV Replication: RNA Quality Also Matters. Viruses, 16(4), 615. https://doi.org/10.3390/v16040615