
Burden, Outcome, and Comorbidities of Extrahepatic Manifestations in Hepatitis B Virus Infections



Abstract
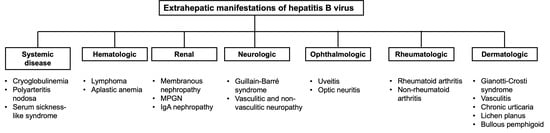
:
1. Introduction
2. Systemic Diseases
2.1. Cryoglobulinemia
2.2. Polyarteritis Nodosa
2.3. Serum Sickness-like Syndrome
3. Hematologic Complications
3.1. Lymphoma
3.2. Aplastic Anemia
4. Renal Disease
4.1. Pathogenesis
4.2. Treatment
4.3. Membranous Nephropathy
4.4. Treatment
4.5. Membranoproliferative Glomerulonephritis
4.6. Treatment
4.7. IgA Nephropathy
5. Neurologic Disease
5.1. Guillain-Barré Syndrome
5.2. Vasculitic Neuropathy and Non-Vasculitic Neuropathy
6. Ophthalmologic Disease
6.1. Uveitis
6.2. Optic Neuritis
7. Rheumatologic Diseases
7.1. Rheumatoid Arthritis
7.2. Non-Rheumatoid Arthritis
7.3. Other Rheumatologic Diseases
8. Dermatologic Manifestations
8.1. Gianotti–Crosti Syndrome or Papular Acrodermatitis of Childhood
8.2. Vasculitis
8.3. Chronic Urticaria and Angioedema
8.4. Lichen Planus
8.5. Bullous Pemphigoid
8.6. Other Cutaneous Manifestations
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kappus, M.R.; Sterling, R.K. Extrahepatic manifestations of acute hepatitis B virus infection. Gastroenterol. Hepatol. 2013, 9, 123–126. [Google Scholar]
- Mazzaro, C.; Adinolfi, L.E.; Pozzato, G.; Nevola, R.; Zanier, A.; Serraino, D.; Andreone, P.; Fenoglio, R.; Sciascia, S.; Gattei, V.; et al. Extrahepatic Manifestations of Chronic HBV Infection and the Role of Antiviral Therapy. J. Clin. Med. 2022, 11, 6247. [Google Scholar] [CrossRef] [PubMed]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef] [PubMed]
- Mason, A. Role of viral replication in extrahepatic syndromes related to hepatitis B virus infection. Minerva Gastroenterol. Dietol. 2006, 52, 53–66. [Google Scholar] [PubMed]
- Mason, A.; Wick, M.; White, H.; Perrillo, R. Hepatitis B virus replication in diverse cell types during chronic hepatitis B virus infection. Hepatology 1993, 18, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.; Theal, J.; Bain, V.; Adams, E.; Perrillo, R. Hepatitis B virus replication in damaged endothelial tissues of patients with extrahepatic disease. Am. J. Gastroenterol. 2005, 100, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Datta, S. Compartmentalization of hepatitis B virus: Looking beyond the liver. World J. Hepatol. 2015, 7, 2241–2244. [Google Scholar] [CrossRef] [PubMed]
- Monti, G.; Galli, M.; Invernizzi, F.; Pioltelli, P.; Saccardo, F.; Monteverde, A.; Pietrogrande, M.; Renoldi, P.; Bombardieri, S.; Bordin, G.; et al. Cryoglobulinaemias: A multi-centre study of the early clinical and laboratory manifestations of primary and secondary disease. GISC. Italian Group for the Study of Cryoglobulinaemias. QJM 1995, 88, 115–126. [Google Scholar] [PubMed]
- Ferri, C.; Sebastiani, M.; Giuggioli, D.; Cazzato, M.; Longombardo, G.; Antonelli, A.; Puccini, R.; Michelassi, C.; Zignego, A.L. Mixed cryoglobulinemia: Demographic, clinical, and serologic features and survival in 231 patients. Semin. Arthritis Rheum. 2004, 33, 355–374. [Google Scholar] [CrossRef]
- Han, H.-x.; Su, W.; Zhou, D.-B.; Li, J.; Cao, X.-x. Hepatitis B virus-related cryoglobulinemia: Clinical characteristics, virological features, and treatment. Virus Res. 2023, 336, 199212. [Google Scholar] [CrossRef]
- Ozkok, A.; Yildiz, A. Hepatitis C virus associated glomerulopathies. World J. Gastroenterol. 2014, 20, 7544–7554. [Google Scholar] [CrossRef] [PubMed]
- Viganò, M.; Martin, P.; Cappelletti, M.; Fabrizi, F. HBV-Associated Cryoglobulinemic Vasculitis: Remission after Antiviral Therapy with Entecavir. Kidney Blood Press. Res. 2014, 39, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Mazzaro, C.; Dal Maso, L.; Gragnani, L.; Visentini, M.; Saccardo, F.; Filippini, D.; Andreone, P.; Zignego, A.L.; Gattei, V.; Monti, G.; et al. Hepatitis B Virus-Related Cryoglobulinemic Vasculitis: Review of the Literature and Long-Term Follow-Up Analysis of 18 Patients Treated with Nucleos(t)ide Analogues from the Italian Study Group of Cryoglobulinemia (GISC). Viruses 2021, 13, 1032. [Google Scholar] [CrossRef]
- Mazzaro, C.; Bomben, R.; Visentini, M.; Gragnani, L.; Quartuccio, L.; Saccardo, F.; Sebastiani, M.; Filippini, D.; Lauletta, G.; Monti, G.; et al. Hepatitis B virus-infection related cryoglobulinemic vasculitis. Clinical manifestations and the effect of antiviral therapy: A review of the literature. Front. Oncol. 2023, 13, 1095780. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Monti, G.; Marson, P.; Scaini, P.; Pietrogrande, M.; Candela, M.; Castelnovo, L.; Faggioli, P.; Novati, P.; Zani, R.; et al. Recommendations for managing the manifestations of severe and life-threatening mixed cryoglobulinemia syndrome. Autoimmun. Rev. 2019, 18, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Jopson, L.; Ng, S.; Lowery, M.; Harwood, J.; Waugh, S.; Valappil, M.; McPherson, S. Improving testing for hepatitis B before treatment with rituximab. Eur. J. Gastroenterol. Hepatol. 2016, 28, 1172–1178. [Google Scholar] [CrossRef]
- Trepo, C.; Guillevin, L.c. Polyarteritis Nodosa and Extrahepatic Manifestations of HBV Infection: The Case Against Autoimmune Intervention in Pathogenesis. J. Autoimmun. 2001, 16, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Lhote, F.; Cohen, P.; Sauvaget, F.; Jarrousse, B.; Lortholary, O.; Noël, L.H.; Trépo, C. Polyarteritis nodosa related to hepatitis B virus. A prospective study with long-term observation of 41 patients. Medicine 1995, 74, 238–253. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Mahr, A.; Cohen, P.; Larroche, C.; Queyrel, V.; Loustaud-Ratti, V.; Imbert, B.; Hausfater, P.; Roudier, J.; Bielefeld, P.; et al. Short-term corticosteroids then lamivudine and plasma exchanges to treat hepatitis B virus-related polyarteritis nodosa. Arthritis Rheum. 2004, 51, 482–487. [Google Scholar] [CrossRef]
- Arkachaisri, T. Serum sickness and hepatitis B vaccine including review of the literature. J. Med. Assoc. Thail. 2002, 85 (Suppl. 2), S607–S612. [Google Scholar]
- Gupta, R.; Fakunle, I.; Samji, V.; Hale, E.B. Serum Sickness-Like Reaction Associated with Acute Hepatitis B in a Previously Vaccinated Adult Male. Cureus 2021, 13, e14742. [Google Scholar] [CrossRef] [PubMed]
- Kuniyoshi, M.; Nakamuta, M.; Sakai, H.; Enjoji, M.; Kinukawa, N.; Kotoh, K.; Fukutomi, M.; Yokota, M.; Nishi, H.; Iwamoto, H.; et al. Prevalence of hepatitis B or C virus infections in patients with non-Hodgkin’s lymphoma. J. Gastroenterol. Hepatol. 2001, 16, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Cucuianu, A.; Patiu, M.; Duma, M.; Basarab, C.; Soritau, O.; Bojan, A.; Vasilache, A.; Mates, M.; Petrov, L. Hepatitis B and C virus infection in Romanian non-Hodgkin’s lymphoma patients. Br. J. Haematol. 1999, 107, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, R.H.; Han, B.; Shi, Y.X.; Luo, H.Y.; Jiang, W.Q.; Lin, T.Y.; Huang, H.Q.; Xia, Z.J.; Guan, Z.Z. High incidence of hepatitis B virus infection in B-cell subtype non-Hodgkin lymphoma compared with other cancers. Cancer 2007, 109, 1360–1364. [Google Scholar] [CrossRef]
- Ulcickas Yood, M.; Quesenberry, C.P., Jr.; Guo, D.; Caldwell, C.; Wells, K.; Shan, J.; Sanders, L.; Skovron, M.L.; Iloeje, U.; Manos, M.M. Incidence of non-Hodgkin’s lymphoma among individuals with chronic hepatitis B virus infection. Hepatology 2007, 46, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Mele, A. Hepatitis viruses and non-Hodgkin lymphoma: Epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011, 117, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, R.H.; Luo, H.Y.; Zhang, D.S.; Jiang, W.Q.; Huang, H.Q.; Sun, X.F.; Xia, Z.J.; Guan, Z.Z. Clinical and prognostic analysis of hepatitis B virus infection in diffuse large B-cell lymphoma. BMC Cancer 2008, 8, 115. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Ye, X.; Su, H.; Li, W.; Liu, D.; Pirmoradian, M.; Wang, X.; Zhang, B.; Zhang, Q.; Chen, L.; et al. Genetic landscape of hepatitis B virus-associated diffuse large B-cell lymphoma. Blood 2018, 131, 2670–2681. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zheng, Y.; Zhu, H.; Lin, X.; Zhang, Y.; Zhao, Y.; Hu, J.; Li, J. Risk of Onset of Hematological Malignancies in Patients Infected with the Hepatitis B Virus: Results from a Large-Scale Retrospective Cohort Study in China. Acta Haematol. 2017, 137, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gan, Y.; Fan, C.; Yuan, H.; Zhang, X.; Shen, Y.; Wang, Q.; Meng, Z.; Xu, D.; Tu, H. Hepatitis B virus and risk of non-Hodgkin lymphoma: An updated meta-analysis of 58 studies. J. Viral Hepat. 2018, 25, 894–903. [Google Scholar] [CrossRef]
- Cao, X.; Wang, Y.; Li, P.; Huang, W.; Lu, X.; Lu, H. HBV Reactivation During the Treatment of Non-Hodgkin Lymphoma and Management Strategies. Front. Oncol. 2021, 11, 685706. [Google Scholar] [CrossRef]
- Zhang, W.; Du, F.; Wang, L.; Bai, T.; Zhou, X.; Mei, H. Hepatitis Virus-associated Non-hodgkin Lymphoma: Pathogenesis and Treatment Strategies. J. Clin. Transl. Hepatol. 2023, 11, 1256–1266. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Pan, S.; Hu, T.; Shen, J.; Zheng, H.; Xie, S.; Xie, Y.; Lu, R.; Guo, L. Capable Infection of Hepatitis B Virus in Diffuse Large B-cell Lymphoma. J. Cancer 2018, 9, 1575–1581. [Google Scholar] [CrossRef]
- Lemaitre, M.; Brice, P.; Frigeni, M.; Hermine, O.; Arcaini, L.; Thieblemont, C.; Besson, C. Hepatitis B virus-associated B-cell non-Hodgkin lymphoma in non-endemic areas in Western Europe: Clinical characteristics and prognosis. J. Infect. 2020, 80, 219–224. [Google Scholar] [CrossRef]
- Brown, K.E.; Tisdale, J.; Barrett, A.J.; Dunbar, C.E.; Young, N.S. Hepatitis-associated aplastic anemia. N. Engl. J. Med. 1997, 336, 1059–1064. [Google Scholar] [CrossRef]
- Alshaibani, A.; Dufour, C.; Risitano, A.; de Latour, R.; Aljurf, M. Hepatitis-Associated Aplastic Anemia. Hematol. Oncol. Stem Cell Ther. 2022, 15, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Casas, R.; Garcia-Buey, L.; Jones, E.A.; Gisbert, J.P.; Moreno-Otero, R. Systematic review: Hepatitis-associated aplastic anaemia—A syndrome associated with abnormal immunological function. Aliment. Pharmacol. Ther. 2009, 30, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Rauff, B.; Idrees, M.; Shah, S.A.; Butt, S.; Butt, A.M.; Ali, L.; Hussain, A.; Irshad Ur, R.; Ali, M. Hepatitis associated aplastic anemia: A review. Virol. J. 2011, 8, 87. [Google Scholar] [CrossRef]
- Foon, K.A.; Mitsuyasu, R.T.; Schroff, R.W.; McIntyre, R.E.; Champlin, R.; Gale, R.P. Immunologic defects in young male patients with hepatitis-associated aplastic anemia. Ann. Intern. Med. 1984, 100, 657–662. [Google Scholar] [CrossRef]
- Ikawa, Y.; Nishimura, R.; Kuroda, R.; Mase, S.; Araki, R.; Maeba, H.; Wada, T.; Toma, T.; Koizumi, S.; Yachie, A. Expansion of a liver-infiltrating cytotoxic T-lymphocyte clone in concert with the development of hepatitis-associated aplastic anaemia. Br. J. Haematol. 2013, 161, 599–602. [Google Scholar] [CrossRef]
- Bowen, D.G.; Warren, A.; Davis, T.; Hoffmann, M.W.; McCaughan, G.W.; Fazekas de St Groth, B.; Bertolino, P. Cytokine-dependent bystander hepatitis due to intrahepatic murine CD8 T-cell activation by bone marrow-derived cells. Gastroenterology 2002, 123, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Kagan, W.A.; Ascensão, J.A.; Pahwa, R.N.; Hansen, J.A.; Goldstein, G.; Valera, E.B.; Incefy, G.S.; Moore, M.A.; Good, R.A. Aplastic anemia: Presence in human bone marrow of cells that suppress myelopoiesis. Proc. Natl. Acad. Sci. USA 1976, 73, 2890–2894. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, C.; Turhan, N.; Yolcu, O.F.; Yilmaz, S. Hepatitis associated with aplastic anemia: Do CD8(+) kupffer cells have a role in the pathogenesis? Dig. Dis. Sci. 2007, 52, 2438–2443. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Bertuch, A.; Sasa, G.S.; Himes, R.W.; Wu, H. Features of Hepatitis in Hepatitis-associated Aplastic Anemia. J. Pediatr. Gastroenterol. Nutr. 2017, 64, e7–e12. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tu, M.; Fu, R.; Wu, Y.; Liu, H.; Xing, L.; Shao, Z. The Clinical and Immune Characteristics of Patients with Hepatitis-Associated Aplastic Anemia in China. PLoS ONE 2014, 9, e98142. [Google Scholar] [CrossRef] [PubMed]
- Paquette, R.L.; Kuramoto, K.; Tran, L.; Sopher, G.; Nimer, S.D.; Zeldis, J.B. Hepatitis C virus infection in acquired aplastic anemia. Am. J. Hematol. 1998, 58, 122–126. [Google Scholar] [CrossRef]
- Mori, T.; Onishi, Y.; Ozawa, Y.; Kato, C.; Kai, T.; Kanda, Y.; Kurokawa, M.; Tanaka, M.; Ashida, T.; Sawayama, Y.; et al. Outcome of allogeneic hematopoietic stem cell transplantation in adult patients with hepatitis-associated aplastic anemia. Int. J. Hematol. 2019, 109, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Locasciulli, A.; Bacigalupo, A.; Bruno, B.; Montante, B.; Marsh, J.; Tichelli, A.; Socié, G.; Passweg, J.; On the Behalf of the Severe Aplastic Anemia Working Party of the European Blood and Marrow Transplant Group; Marrow Transplant, G. Hepatitis-associated aplastic anaemia: Epidemiology and treatment results obtained in Europe. A report of The EBMT aplastic anaemia working party. Br. J. Haematol. 2010, 149, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Combes, B.; Shorey, J.; Barrera, A.; Stastny, P.; Eigenbrodt, E.H.; Hull, A.R.; Carter, N.W. Glomerulonephritis with deposition of Australia antigen-antibody complexes in glomerular basement membrane. Lancet 1971, 2, 234–237. [Google Scholar] [CrossRef]
- Levy, M.; Chen, N. Worldwide perspective of hepatitis B-associated glomerulonephritis in the 80s. Kidney Int. Suppl. 1991, 35, S24–S33. [Google Scholar]
- Lai, K.N.; Li, P.K.; Lui, S.F.; Au, T.C.; Tam, J.S.; Tong, K.L.; Lai, F.M. Membranous nephropathy related to hepatitis B virus in adults. N. Engl. J. Med. 1991, 324, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.M. Hepatitis B and Renal Disease. Curr. Hepat. Rep. 2010, 9, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Diao, Z.; Ding, J.; Yin, C.; Wang, L.; Liu, W. Purified hepatitis B virus induces human Mesangial cell proliferation and extracellular matrix expression In Vitro. Virol. J. 2013, 10, 300. [Google Scholar] [CrossRef]
- Jiang, W.; Liu, T.; Dong, H.; Xu, Y.; Liu, L.Q.; Guan, G.J.; Liu, X.C. Relationship Between Serum DNA Replication, Clinicopathological Characteristics and Prognosis of Hepatitis B Virus-associated Glomerulonephritis with Severe Proteinuria by Lamivudine Plus Adefovir Dipivoxil Combination Therapy. Biomed. Environ. Sci. 2015, 28, 206–213. [Google Scholar] [CrossRef]
- Pyrsopoulos, N.T.; Reddy, K.R. Extrahepatic manifestations of chronic viral hepatitis. Curr. Gastroenterol. Rep. 2001, 3, 71–78. [Google Scholar] [CrossRef]
- Shah, A.S.; Amarapurkar, D.N. Spectrum of hepatitis B and renal involvement. Liver Int. Off. J. Int. Assoc. Study Liver 2018, 38, 23–32. [Google Scholar] [CrossRef]
- Wang, L.; Ye, Z.; Liang, H.; Zhang, B.; Xu, L.; Feng, Z.; Liu, S.; Shi, W. The combination of tacrolimus and entecavir improves the remission of HBV-associated glomerulonephritis without enhancing viral replication. Am. J. Transl. Res. 2016, 8, 1593–1600. [Google Scholar] [CrossRef]
- Wang, L.; Xie, B.; Zheng, Q.; Xu, L.; Ye, Z. Efficacy of entecavir in treating hepatitis B virus-associated membranous nephropathy. Rev. Esp. Enferm. Dig. 2020, 112, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wu, Y.; Zheng, B.; Zhang, X.; An, D.; Guo, N.; Wang, J.; Guo, Y.; Tang, L. Clinicopathological characteristics and prognosis of hepatitis B associated membranous nephropathy and idiopathic membranous nephropathy complicated with hepatitis B virus infection. Sci. Rep. 2021, 11, 18407. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, Y.P.; Chen, D.P.; Zhuo, L.; Li, W.G. A Meta-Analysis of Antiviral Therapy for Hepatitis B Virus-Associated Membranous Nephropathy. PLoS ONE 2016, 11, e0160437. [Google Scholar] [CrossRef]
- Moroni, G.; Ponticelli, C. Secondary Membranous Nephropathy. A Narrative Review. Front. Med. 2020, 7, 611317. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.D.; Wiggelinkhuizen, J. The clinical course of hepatitis B virus-associated nephropathy. Pediatr. Nephrol. 1994, 8, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.C.; Wu, C.Y.; Lin, C.Y.; Lin, G.J.; Chen, C.H.; Huang, F.Y. Membranous nephropathy in 52 hepatitis B surface antigen (HBsAg) carrier children in Taiwan. Kidney Int. 1989, 36, 1103–1107. [Google Scholar] [CrossRef]
- Lin, C.Y. Treatment of hepatitis B virus-associated membranous nephropathy with recombinant alpha-interferon. Kidney Int. 1995, 47, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R. Clinical presentation & management of glomerular diseases: Hematuria, nephritic & nephrotic syndrome. Mo. Med. 2011, 108, 33–36. [Google Scholar] [PubMed]
- Alchi, B.; Jayne, D. Membranoproliferative glomerulonephritis. Pediatr. Nephrol. 2010, 25, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Bhimma, R.; Coovadia, H.M. Hepatitis B virus-associated nephropathy. Am. J. Nephrol. 2004, 24, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Mochizuki, T.; Akihisa, T.; Kawasoe, K.; Kawachi, K.; Makabe, S.; Sawada, A.; Manabe, S.; Sato, M.; Amemiya, N.; et al. Successful entecavir plus prednisolone treatment for hepatitis B virus-associated membranoproliferative glomerulonephritis: A case report. Medicine 2019, 98, e14014. [Google Scholar] [CrossRef]
- Rodrigues, J.C.; Haas, M.; Reich, H.N. IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 677–686. [Google Scholar] [CrossRef]
- McGrogan, A.; Franssen, C.F.; de Vries, C.S. The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol. Dial. Transplant. 2011, 26, 414–430. [Google Scholar] [CrossRef]
- Lai, K.N.; Lai, F.M.; Tam, J.S.; Vallance-Owen, J. Strong association between IgA nephropathy and hepatitis B surface antigenemia in endemic areas. Clin. Nephrol. 1988, 29, 229–234. [Google Scholar] [PubMed]
- Zhang, Y.E.; Guo, M.Y.; Ying, Y.Y. Further study on the immunopathology of hepatitis B virus associated glomerulonephritis. Zhonghua Nei Ke Za Zhi 1990, 29, 526–529, 574. [Google Scholar] [PubMed]
- Wang, N.S.; Wu, Z.L.; Zhang, Y.E.; Guo, M.Y.; Liao, L.T. Role of hepatitis B virus infection in pathogenesis of IgA nephropathy. World J. Gastroenterol. 2003, 9, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N.; Tam, J.S.; Lin, H.J.; Lai, F.M. The therapeutic dilemma of the usage of corticosteroid in patients with membranous nephropathy and persistent hepatitis B virus surface antigenaemia. Nephron 1990, 54, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Miyazaki, J.; Hino, O.; Tomita, N.; Chisaka, O.; Matsubara, K.; Yamamura, K. Expression and replication of hepatitis B virus genome in transgenic mice. Proc. Natl. Acad. Sci. USA 1989, 86, 207–211. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G. Natural history of idiopathic IgA nephropathy: Role of clinical and histological prognostic factors. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2000, 36, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yang, K.; Xiao, B.; Lin, J.; Zhao, Q.; Bhuva, M.S.; Yang, H. Efficacy and safety of angiotensin-converting enzyme inhibitors/angiotensin receptor blocker therapy for IgA nephropathy: A meta-analysis of randomized controlled trials. J. Cell Biochem. 2019, 120, 3689–3695. [Google Scholar] [CrossRef]
- Nagasawa, Y.; Yamamoto, R.; Shinzawa, M.; Shoji, T.; Hasuike, Y.; Nagatoya, K.; Yamauchi, A.; Hayashi, T.; Kuragano, T.; Moriyama, T.; et al. Efficacy of corticosteroid therapy for IgA nephropathy patients stratified by kidney function and proteinuria. Clin. Exp. Nephrol. 2020, 24, 927–934. [Google Scholar] [CrossRef]
- Fang, J.; Li, W.; Tan, Z.; Li, D. Comparison of prednisolone and lamivudine combined therapy with prednisolone monotherapy on carriers of hepatitis B virus with IgA nephropathy: A prospective cohort study. Int. Urol. Nephrol. 2014, 46, 49–56. [Google Scholar] [CrossRef]
- Poropatich, K.O.; Walker, C.L.; Black, R.E. Quantifying the association between Campylobacter infection and Guillain-Barré syndrome: A systematic review. J. Health Popul. Nutr. 2010, 28, 545–552. [Google Scholar] [CrossRef]
- Yuki, N.; Hartung, H.P. Guillain-Barré syndrome. N. Engl. J. Med. 2012, 366, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Duan, S. Severe Guillain-Barré syndrome associated with chronic hepatitis B: A case report and literature review. Medicine 2021, 100, e27989. [Google Scholar] [CrossRef] [PubMed]
- Yimam, K.; Frederick, T.; Merriman, R.; Talavera, J. Acute Hepatitis B Infection Causing Guillain-Barré Syndrome (GBS): A Rare Case: 2011 ACG Presidential Poster: 798. Off. J. Am. Coll. Gastroenterol. ACG 2011, 106, S299–S300. [Google Scholar] [CrossRef]
- Tsukada, N.; Koh, C.S.; Inoue, A.; Yanagisawa, N. Demyelinating neuropathy associated with hepatitis B virus infection. Detection of immune complexes composed of hepatitis B virus surface antigen. J. Neurol. Sci. 1987, 77, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Khamaisi, M.; Shoenfeld, Y.; Orbach, H. Guillain-Barré syndrome following hepatitis B vaccination. Clin. Exp. Rheumatol. 2004, 22, 767–770. [Google Scholar] [PubMed]
- Burns, T.M.; Schaublin, G.A.; Dyck, P.J. Vasculitic neuropathies. Neurol. Clin. 2007, 25, 89–113. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.P.; Dyck, P.J.; Gronseth, G.S.; Guillevin, L.; Hadden, R.D.; Heuss, D.; Léger, J.M.; Notermans, N.C.; Pollard, J.D.; Said, G.; et al. Peripheral Nerve Society Guideline on the classification, diagnosis, investigation, and immunosuppressive therapy of non-systemic vasculitic neuropathy: Executive summary. J. Peripher. Nerv. Syst. 2010, 15, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, N.; Koh, C.S.; Owa, M.; Yanagisawa, N. Chronic neuropathy associated with immune complexes of hepatitis B virus. J. Neurol. Sci. 1983, 61, 193–210. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, K. Hepatotropic viral infection associated systemic vasculitides-hepatitis B virus associated polyarteritis nodosa and hepatitis C virus associated cryoglobulinemic vasculitis. J. Clin. Exp. Hepatol. 2013, 3, 204–212. [Google Scholar] [CrossRef]
- Kusama, K.; Nakae, Y.; Tada, M.; Higashiyama, Y.; Miyaji, Y.; Yamaura, G.; Kunii, M.; Tanaka, K.; Ohyama, K.; Koike, H.; et al. Hepatitis B Virus-related Vasculitic Neuropathy in an Inactive Virus Carrier Treated with Intravenous Immunoglobulin. Intern. Med. 2020, 59, 3075–3078. [Google Scholar] [CrossRef] [PubMed]
- Olney, R.K. AAEM minimonograph #38: Neuropathies in connective tissue disease. Muscle Nerve 1992, 15, 531–542. [Google Scholar] [CrossRef]
- Lacomis, D.; Zivković, S.A. Approach to vasculitic neuropathies. J. Clin. Neuromuscul. Dis. 2007, 9, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Nam, T.S.; Lee, S.H.; Park, M.S.; Choi, K.H.; Kim, J.T.; Choi, S.M.; Kim, B.C.; Kim, M.K.; Cho, K.H. Mononeuropathy multiplex in a patient with chronic active hepatitis B. J. Clin. Neurol. 2010, 6, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.C.; Gowda, V.N.; Panda, P.K. Hepatitis B-Related Mononeuritis Multiplex. Am. J. Infect. Dis. 2022, 18, 54–57. [Google Scholar] [CrossRef]
- Pasquet, F.; Combarnous, F.; Macgregor, B.; Coppere, B.; Mausservey, C.; Ninet, J.; Hot, A. Safety and efficacy of rituximab treatment for vasculitis in hepatitis B virus-associated type II cryoglobulinemia: A case report. J. Med. Case Rep. 2012, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Terrier, B.; Marie, I.; Lacraz, A.; Belenotti, P.; Bonnet, F.; Chiche, L.; Graffin, B.; Hot, A.; Kahn, J.E.; Michel, C.; et al. Non HCV-related infectious cryoglobulinemia vasculitis: Results from the French nationwide CryoVas survey and systematic review of the literature. J. Autoimmun. 2015, 65, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.H.; Ilyas, K.; Ghazanfar, H.; Khan, H.H.; Hussain, Q.; Hammad, S.; Munir, A.; Asim, R. Fatal Fulminant Hepatitis from Rituximab-induced Hepatitis B Reactivation in a Patient with Follicular Lymphoma: A Case Report and a Brief Review of Literature. Cureus 2018, 10, e2257. [Google Scholar] [CrossRef] [PubMed]
- Bodaghi, B.; Cassoux, N.; Wechsler, B.; Hannouche, D.; Fardeau, C.; Papo, T.; Huong, D.L.; Piette, J.C.; LeHoang, P. Chronic severe uveitis: Etiology and visual outcome in 927 patients from a single center. Medicine 2001, 80, 263–270. [Google Scholar] [CrossRef]
- Tien, P.T.; Lin, C.J.; Tsai, Y.Y.; Chen, H.S.; Hwang, D.K.; Muo, C.H.; Lin, J.M.; Chen, W.L. Relationship between uveitis, different types of viral hepatitis, and liver cirrhosis: A 12-Year Nationwide Population-Based Cohort Study. Retina 2016, 36, 2391–2398. [Google Scholar] [CrossRef]
- Grob, P.J.; Martenet, A.C.; Witmer, R. Nonspecific immune parameters and hepatitis B antigens in patients with uveitis. Mod. Probl. Ophthalmol. 1976, 16, 254–258. [Google Scholar] [PubMed]
- Fraunfelder, F.W.; Suhler, E.B.; Fraunfelder, F.T. Hepatitis B vaccine and uveitis: An emerging hypothesis suggested by review of 32 case reports. Cutan. Ocul. Toxicol. 2010, 29, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Köksal, I.; Cetinkaya, K.; Aker, F. Hepatitis B surface antigen in tears and aqueous humor. A comparative study of serum hepatitis B surface antigen levels. Ophthalmologica 1992, 204, 19–22. [Google Scholar] [CrossRef]
- Singh, D.P.; Kikuchi, T.; Singh, V.K.; Shinohara, T. A single amino acid substitution in core residues of S-antigen prevents experimental autoimmune uveitis. J. Immunol. 1994, 152, 4699–4705. [Google Scholar] [CrossRef]
- Maya, R.; Gershwin, M.E.; Shoenfeld, Y. Hepatitis B virus (HBV) and autoimmune disease. Clin. Rev. Allergy Immunol. 2008, 34, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Zhong, Z.; Su, G.; Feng, X.; Du, L.; Li, F.Z.; Dai, L.; Kijlstra, A.; Yang, P. Surveillance of Liver Function in Uveitis with or without Chronic HBV Infection. Ophthalmic Res. 2022, 65, 94–103. [Google Scholar] [CrossRef]
- Redenbaugh, V.; Flanagan, E.P. Monoclonal Antibody Therapies Beyond Complement for NMOSD and MOGAD. Neurotherapeutics 2022, 19, 808–822. [Google Scholar] [CrossRef]
- Achiron, L.R. Postinfectious hepatitis B optic neuritis. Optom. Vis. Sci. 1994, 71, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Heekin, R.; Gandhy, C.; Robertson, D. Seronegative Neuromyelitis Optica Spectrum Disorder following Exposure to Hepatitis B Vaccination. Case Rep. Neurol. 2015, 7, 78–83. [Google Scholar] [CrossRef]
- Wands, J.R.; Mann, E.; Alpert, E.; Isselbacher, K.J. The pathogenesis of arthritis associated with acute hepatitis-B surface antigen-positive hepatitis. Complement activation and characterization of circulating immune complexes. J. Clin. Investig. 1975, 55, 930–936. [Google Scholar] [CrossRef]
- Baig, S.; Alamgir, M. The extrahepatic manifestations of hepatitis B virus. J. Coll. Physicians Surg. Pak. 2008, 18, 451–457. [Google Scholar] [PubMed]
- Molina, V.; Shoenfeld, Y. Infection, vaccines and other environmental triggers of autoimmunity. Autoimmunity 2005, 38, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, Í.M.X.; Silva, R. Rheumatological Manifestations Associated with Viral Hepatitis B or C. Rev. Soc. Bras. Med. Trop. 2019, 52, e20180407. [Google Scholar] [CrossRef]
- Csepregi, A.; Nemesanszky, E.; Rojkovich, B.; Poor, G. Rheumatoid arthritis and hepatitis B virus: Evaluating the pathogenic link. J. Rheumatol. 2001, 28, 474–477. [Google Scholar] [PubMed]
- Li, S.; Yu, Y.; Yue, Y.; Zhang, Z.; Su, K. Microbial Infection and Rheumatoid Arthritis. J. Clin. Cell Immunol. 2013, 4, 174. [Google Scholar] [CrossRef] [PubMed]
- Franssila, R.; Hedman, K. Infection and musculoskeletal conditions: Viral causes of arthritis. Best. Pract. Res. Clin. Rheumatol. 2006, 20, 1139–1157. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.I.; Yoo, W.H.; Yun, H.J.; Kim, D.S.; Lee, H.S.; Choi, S.I.; Hur, J.A.; Cho, Y.G. Absence of antibody to cyclic citrullinated peptide in sera of non-arthritic patients with chronic hepatitis B virus infection. Clin. Rheumatol. 2007, 26, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.K.; Sheen, D.H.; Lee, Y.J.; Mun, Y.R.; Park, M.; Shim, S.C. Anti-cyclic citrullinated peptide antibodies distinguish hepatitis B virus (HBV)-associated arthropathy from concomitant rheumatoid arthritis in patients with chronic HBV infection. J. Rheumatol. 2009, 36, 712–716. [Google Scholar] [CrossRef]
- Senna, E.R.; De Barros, A.L.; Silva, E.O.; Costa, I.F.; Pereira, L.V.; Ciconelli, R.M.; Ferraz, M.B. Prevalence of rheumatic diseases in Brazil: A study using the COPCORD approach. J. Rheumatol. 2004, 31, 594–597. [Google Scholar]
- Feuchtenberger, M.; Schäfer, A.; Philipp Nigg, A.; Rupert Kraus, M. Hepatitis B Serology in Patients with Rheumatic Diseases. Open Rheumatol. J. 2016, 10, 39–48. [Google Scholar] [CrossRef]
- Hsu, C.S.; Lang, H.C.; Huang, K.Y.; Lin, H.H.; Chen, C.L. Association of Rheumatoid Arthritis and Hepatitis B Infection: A Nationwide Nested Case-Control Study from 1999 to 2009 in Taiwan. Medince 2016, 95, e3551. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.J.; Zhu, L.J.; Li, Y.H.; Mo, Y.Q.; Zheng, D.H.; Ma, J.D.; Ou-Yang, X.; Pessler, F.; Dai, L. The association between hepatitis B virus infection and disease activity, synovitis, or joint destruction in rheumatoid arthritis. Clin. Rheumatol. 2013, 32, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Cacoub, P.; Terrier, B. Hepatitis B-related autoimmune manifestations. Rheum. Dis. Clin. N. Am. 2009, 35, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Ozsahin, M.; Gonen, I.; Ermis, F.; Oktay, M.; Besir, F.H.; Kutlucan, A.; Sahin, A.; Ataoglu, S. The prevalence of fibromyalgia among patients with hepatitis B virus infection. Int. J. Clin. Exp. Med. 2013, 6, 804–808. [Google Scholar] [PubMed]
- Vassilopoulos, D.; Calabrese, L.H. Virally associated arthritis 2008: Clinical, epidemiologic, and pathophysiologic considerations. Arthritis Res. Ther. 2008, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Inman, R.D. Rheumatic manifestations of hepatitis B virus infection. Semin. Arthritis Rheum. 1982, 11, 406–420. [Google Scholar] [CrossRef]
- Maslennikov, R.; Ivashkin, V.; Efremova, I.; Shirokova, E. Immune disorders and rheumatologic manifestations of viral hepatitis. World J. Gastroenterol. 2021, 27, 2073–2089. [Google Scholar] [CrossRef]
- Cacoub, P.; Asselah, T. Hepatitis B Virus Infection and Extra-Hepatic Manifestations: A Systemic Disease. Am. J. Gastroenterol. 2022, 117, 253–263. [Google Scholar] [CrossRef]
- Han, S.H. Extrahepatic manifestations of chronic hepatitis B. Clin. Liver Dis. 2004, 8, 403–418. [Google Scholar] [CrossRef]
- Alpert, E.; Schur, P.H.; Isselbacher, K.J. Sequential changes of serum complement in HAA related arthritis. N. Engl. J. Med. 1972, 287, 103. [Google Scholar] [CrossRef]
- Duffy, J.; Lidsky, M.D.; Sharp, J.T.; Davis, J.S.; Person, D.A.; Hollinger, F.B.; Min, K.W. Polyarthritis, polyarteritis and hepatitis B. Medince 1976, 55, 19–37. [Google Scholar] [CrossRef]
- Yazmalar, L.; Deveci, Ö.; Batmaz, İ.; İpek, D.; Çelepkolu, T.; Alpaycı, M.; Hattapoğlu, E.; Akdeniz, D.; Sarıyıldız, M.A. Fibromyalgia incidence among patients with hepatitis B infection. Int. J. Rheum. Dis. 2016, 19, 637–643. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Xu, X.; Hu, W.; Shen, H.; Chen, J. Prevalence of hepatitis B virus and hepatitis C virus infection in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Oncotarget 2017, 8, 102437–102445. [Google Scholar] [CrossRef]
- Zandman-Goddard, G.; Shoenfeld, Y. Infections and SLE. Autoimmunity 2005, 38, 473–485. [Google Scholar] [CrossRef]
- Looi, L.M.; Prathap, K. Hepatitis B virus surface antigen in glomerular immune complex deposits of patients with systemic lupus erythematosus. Histopathology 1982, 6, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Dienstag, J.L. Immunopathogenesis of the extrahepatic manifestations of hepatitis B virus infection. Springer Semin. Immunopathol. 1981, 3, 461–472. [Google Scholar] [CrossRef]
- Vassilopoulos, D.; Manolakopoulos, S. Rheumatic manifestations of hepatitis. Curr. Opin. Rheumatol. 2010, 22, 91–96. [Google Scholar] [CrossRef]
- Hu, P.; Ren, H. Interpretations of EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. Zhonghua Gan Zang Bing Za Zhi 2017, 25, 415–418. [Google Scholar] [CrossRef]
- Luo, S.Q.; Zhang, L.X. AASLD practice guidelines: Chronic hepatitis B. Zhonghua Gan Zang Bing Za Zhi 2007, 15, 477–480. [Google Scholar] [PubMed]
- Parsons, M.E.; Russo, G.G.; Millikan, L.E. Dermatologic disorders associated with viral hepatitis infections. Int. J. Dermatol. 1996, 35, 77–81. [Google Scholar] [CrossRef]
- McElgunn, P.S. Dermatologic manifestations of hepatitis B virus infection. J. Am. Acad. Dermatol. 1983, 8, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Kochhar, A.M.; Reddy, B.S.N. Cutaneous manifestations of hepatitis b and c virus infections: A study of 100 cases. Indian. J. Dermatol. 2003, 48, 73–77. [Google Scholar]
- Popp, J.W., Jr.; Harrist, T.J.; Dienstag, J.L.; Bhan, A.K.; Wands, J.R.; LaMont, J.T.; Mihm, M.C., Jr. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch. Intern. Med. 1981, 141, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Akhter, A.; Said, A. Cutaneous manifestations of viral hepatitis. Curr. Infect. Dis. Rep. 2015, 17, 452. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M.; Warken, K.; Tyring, S.K. The cutaneous manifestations of viral hepatitis. Dermatol. Clin. 2002, 20, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Cozzani, E.; Herzum, A.; Burlando, M.; Parodi, A. Cutaneous manifestations of HAV, HBV, HCV. Ital. J. Dermatol. Venerol. 2021, 156, 5–12. [Google Scholar] [CrossRef]
- Berant, M.; Naveh, Y.; Weissman, I. Papular acrodermatitis with cytomegalovirus hepatitis. Arch. Dis. Child. 1983, 58, 1024–1025. [Google Scholar] [CrossRef] [PubMed]
- Gocke, D.J. Extrahepatic manifestations of viral hepatitis. Am. J. Med. Sci. 1975, 270, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Lockshin, N.A.; Hurley, H. Urticaria as a sign of viral hepatitis. Arch. Dermatol. 1972, 105, 570–571. [Google Scholar] [CrossRef]
- Kolkhir, P.; Pereverzina, N.; Olisova, O.; Maurer, M. Comorbidity of viral hepatitis and chronic spontaneous urticaria: A systematic review. Allergy 2018, 73, 1946–1953. [Google Scholar] [CrossRef]
- Dogan, B. Dermatological manifestations in hepatitis B surface antigen carriers in east region of Turkey. J. Eur. Acad. Dermatol. Venereol. 2005, 19, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Rebora, A.; Rongioletti, F. Lichen planus and chronic active hepatitis. J. Am. Acad. Dermatol. 1984, 10, 840–841. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Pisanti, S. Oral erosive lichen planus and chronic active hepatitis. J. Am. Acad. Dermatol. 1985, 12, 719. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, Z.; Ji, X.; Su, S.; Liu, X.; Xu, S.; Han, Y.; Mu, D.; Liu, H. Lack of evidence of hepatitis in patients with oral lichen planus in China: A case control study. Med. Oral Patol. Oral Cir. Bucal 2016, 21, e161–e168. [Google Scholar] [CrossRef] [PubMed]
- El-Rifaei, A.M.; Fathalla, S.E.; Al-Sheikh, I.H.; Tinguria, M.B.; Qadry, Y.A. The prevalence of indices of hepatitis C and B infection, and elevated aminotransferase enzymes in patients with oral lichen planus (olp) in eastern saudi arabia. J. Fam. Community Med. 1998, 5, 39–43. [Google Scholar]
- Wang, J.H.; Hung, S.J. Lichen planus associated with hepatitis B, hepatitis C, and liver cirrhosis in a nationwide cohort study. J. Am. Acad. Dermatol. 2021, 84, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Baykal, C.; Okan, G.; Sarica, R. Childhood bullous pemphigoid developed after the first vaccination. J. Am. Acad. Dermatol. 2001, 44, 348–350. [Google Scholar] [CrossRef]
- Grigorescu, I.; Dumitrascu, D.L. Spontaneous and antiviral-induced cutaneous lesions in chronic hepatitis B virus infection. World J. Gastroenterol. 2014, 20, 15860–15866. [Google Scholar] [CrossRef] [PubMed]
- Chevrant-Breton, J.; Logeais, B.; Pibouin, M. Pyoderma gangrenosum (phagedenic pyoderma). Ann. Dermatol. Venereol. 1989, 116, 577–589. [Google Scholar] [PubMed]
- Cervia, M.; Parodi, A.; Rebora, A. Chronic active hepatitis and erythema nodosum. Arch. Dermatol. 1982, 118, 878. [Google Scholar] [CrossRef]

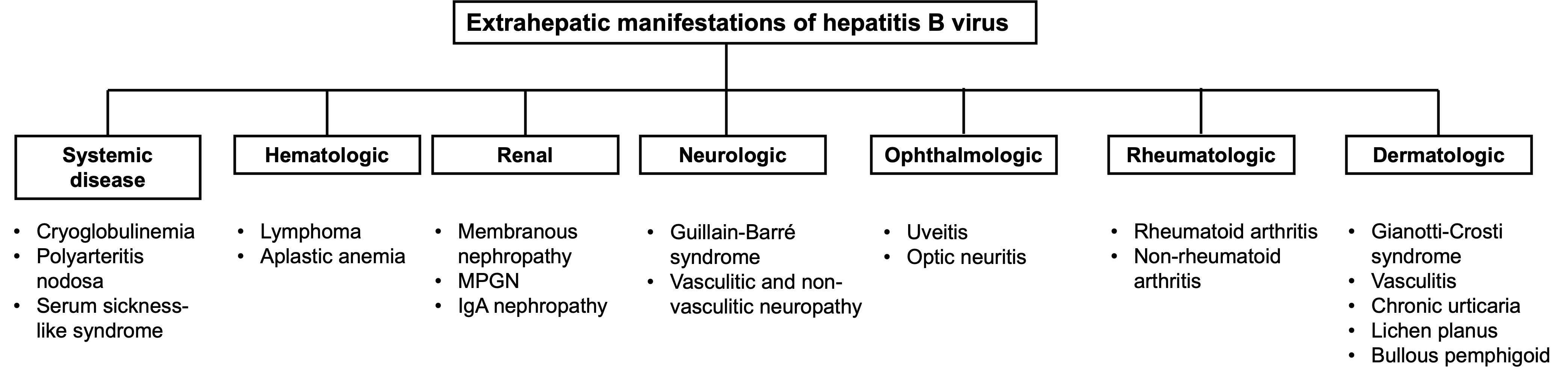{kind=link}
| Extrahepatic Manifestations | Pathophysiology | Treatment |
|---|---|---|
| Systemic diseases Cryoglobulinemia | Circulating immune complexes and chronic antigen stimulation |
|
| Polyarteritis nodosa | Immune dysregulation and immune complexes deposition | Antiviral therapy In severe cases, antiviral therapy and glucocorticoids ± plasma exchange |
| Serum sickness-like syndrome | Immune complexes lead to consumption of complement | Supportive treatment |
| Hematologic Lymphoma (NHL) |
| Antiviral therapy and immunotherapy |
| Aplastic anemia | Bone marrow destruction by clonal expansion of cytotoxic T cells |
|
| Renal Membranous nephropathy | Immune complex (HBsAg and Ab) deposition in the subepithelial space | Antiviral therapy, immunosuppressive |
| Membranoproliferative glomerulonephritis | Immune complex deposition in the mesangium | Not well established- a case report used antiviral therapy and glucocorticoids |
| IgA nephropathy | Viral transcription in the glomerular and renal tubular epithelial cells | Antiviral and immunosuppressive therapy |
| Neurologic Guillain-Barré Syndrome | Not well understood | Standard treatment with IVIG and antiviral therapy |
| Vasculitic neuropathy and non-vasculitic neuropathy | Immune complexes deposition in the vasa nervorum’s endothelium | Antiviral therapy, glucocorticoids, and rituximab |
| Ophthalmic Uveitis | Complement mediated immune process in the retinal vasculature | Antiviral therapy and supportive treatment |
| Optic neuritis | Not well understood | Not enough studies |
| Rheumatologic Rheumatoid arthritis | Immune complex deposition and production of autoantibodies | Antiviral therapy before immunosuppressive therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Songtanin, B.; Chaisrimaneepan, N.; Mendóza, R.; Nugent, K. Burden, Outcome, and Comorbidities of Extrahepatic Manifestations in Hepatitis B Virus Infections. Viruses 2024, 16, 618. https://doi.org/10.3390/v16040618
Songtanin B, Chaisrimaneepan N, Mendóza R, Nugent K. Burden, Outcome, and Comorbidities of Extrahepatic Manifestations in Hepatitis B Virus Infections. Viruses. 2024; 16(4):618. https://doi.org/10.3390/v16040618
Chicago/Turabian StyleSongtanin, Busara, Nattanicha Chaisrimaneepan, Roberto Mendóza, and Kenneth Nugent. 2024. "Burden, Outcome, and Comorbidities of Extrahepatic Manifestations in Hepatitis B Virus Infections" Viruses 16, no. 4: 618. https://doi.org/10.3390/v16040618
APA StyleSongtanin, B., Chaisrimaneepan, N., Mendóza, R., & Nugent, K. (2024). Burden, Outcome, and Comorbidities of Extrahepatic Manifestations in Hepatitis B Virus Infections. Viruses, 16(4), 618. https://doi.org/10.3390/v16040618