
Intracellular “In Silico Microscopes”—Comprehensive 3D Spatio-Temporal Virus Replication Model Simulations


Abstract
1. Introduction
1.1. Biological Basics
1.2. Aims of this Study
1.3. Organization of this Paper
2. Materials, Models and Methods
2.1. Computational Domain: Experimental Data Based Unstructured Grid Geometry
2.2. Tangential Space and Manifold Differential Operators
2.3. Mathematical Partial Differential Equation (PDE) Model Coupling Surface and Volume Effects
2.4. Michaelis–Menten Kinetics Inspired Reaction Terms
2.5. Numerical Values of the Model Parameters
2.6. Numerical Solution Techniques for Solving the sufPDE/PDE System
2.7. Integrals of Concentrations over Subdomains
2.8. Distinguishing between Geometrically Defined and Biophysically Active MW Zones
3. Results
3.1. Geometric Basis and Geometric versus Biophysical Subdomains
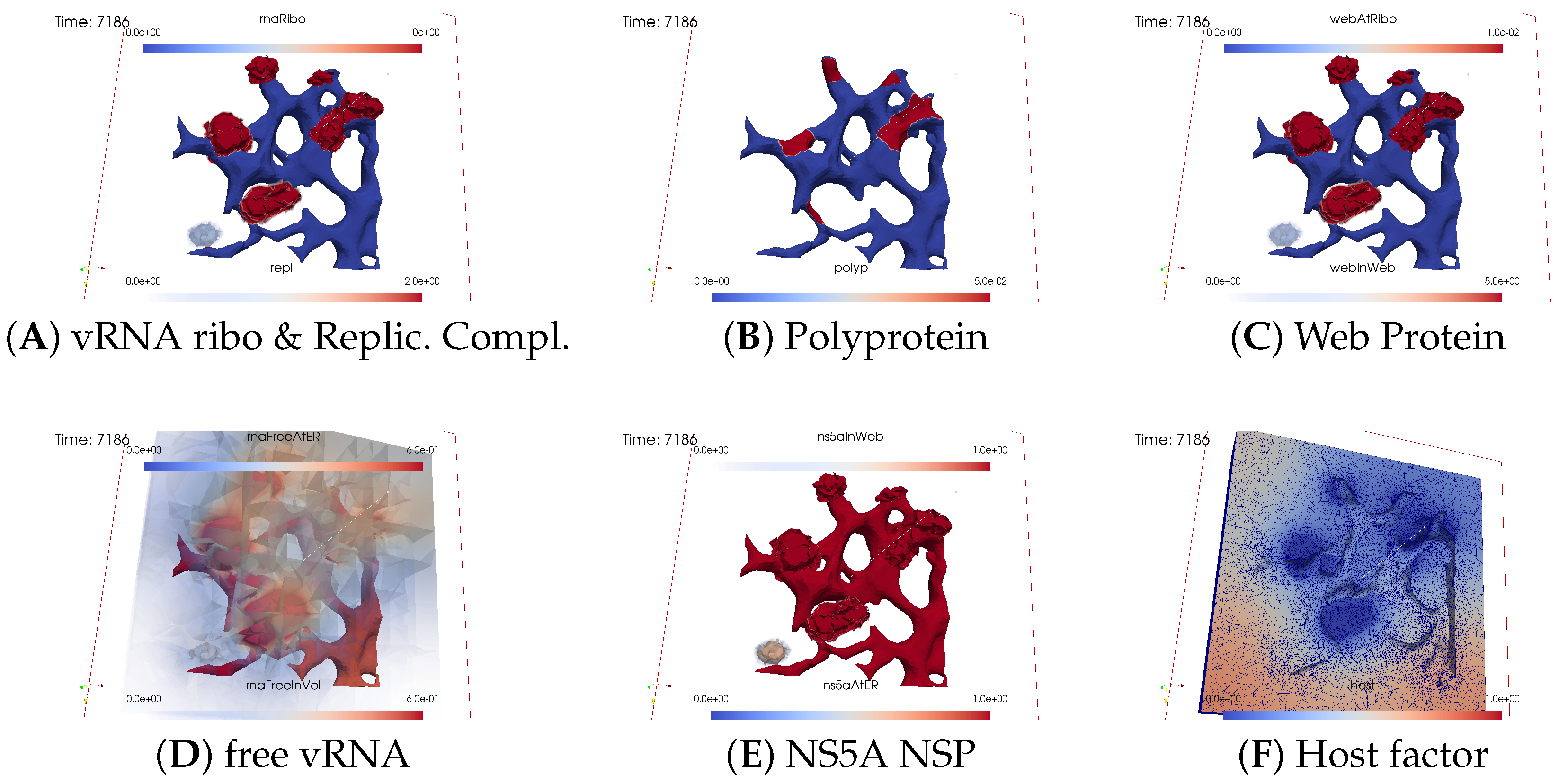
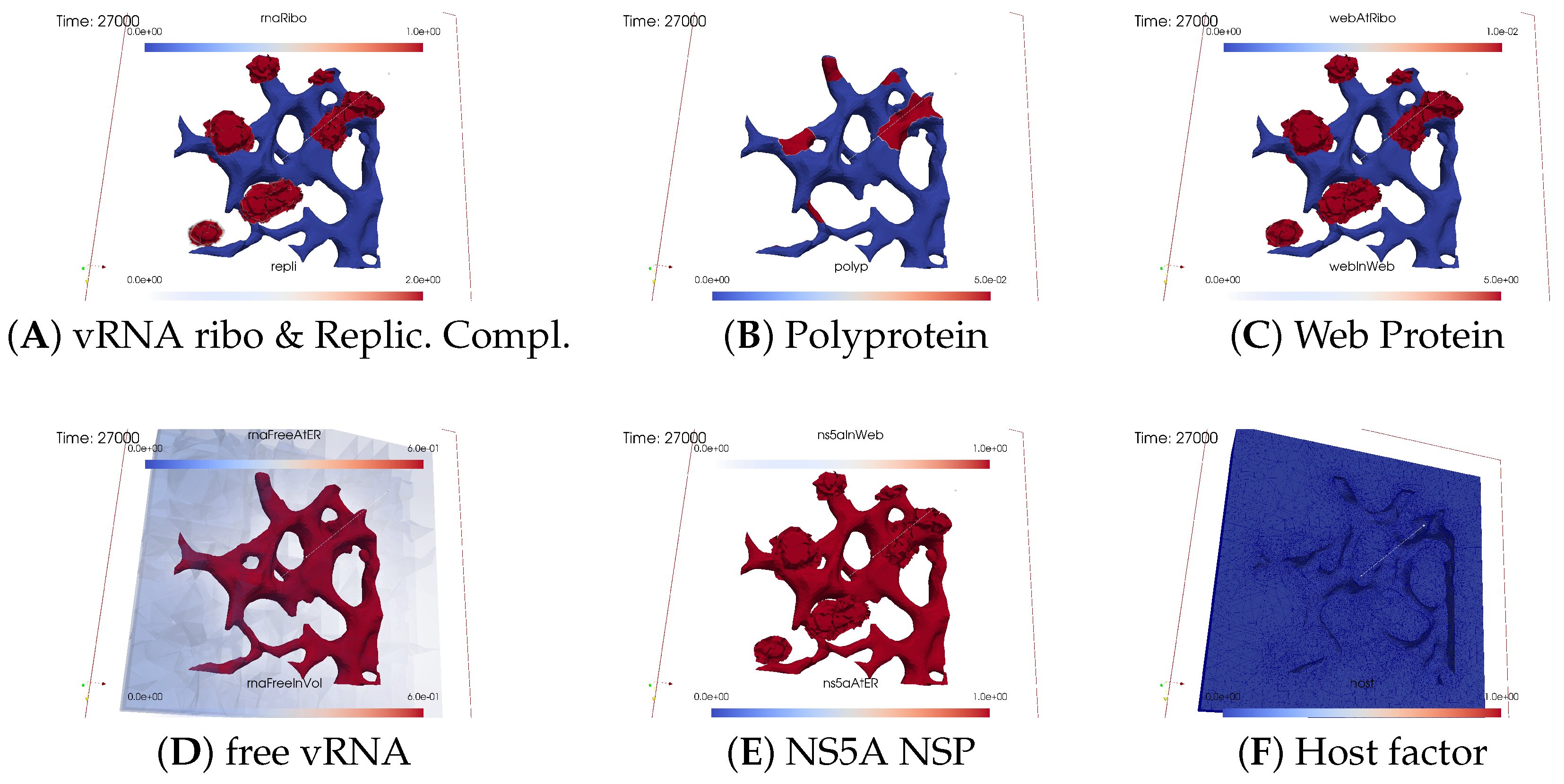
3.2. Spatial Simulation Data Evaluation—Simulation Movie
3.2.1. Concentrations and Their Spatial Regions
3.2.2. In Silico Microscopye of the vRNA Cycle
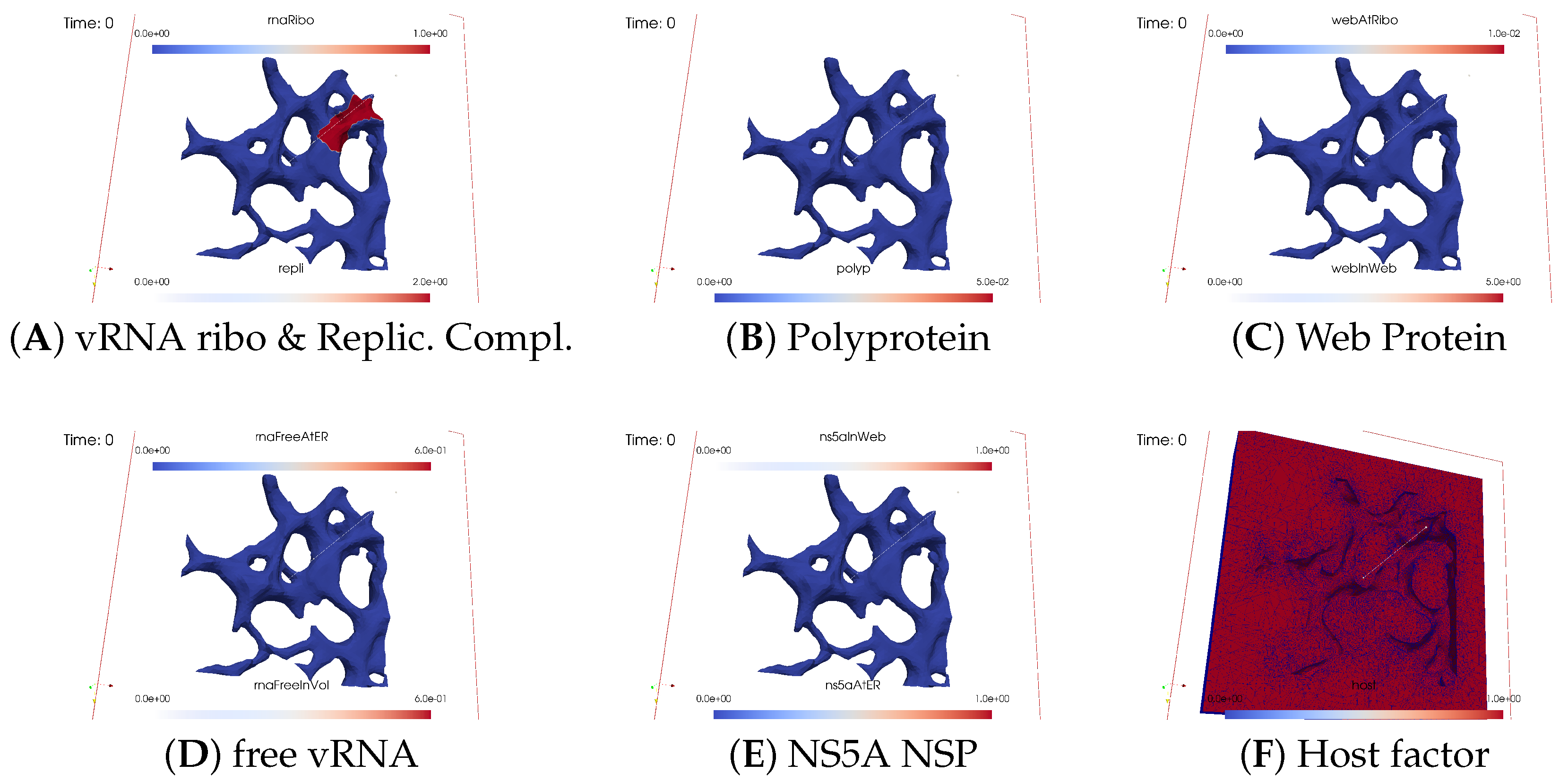
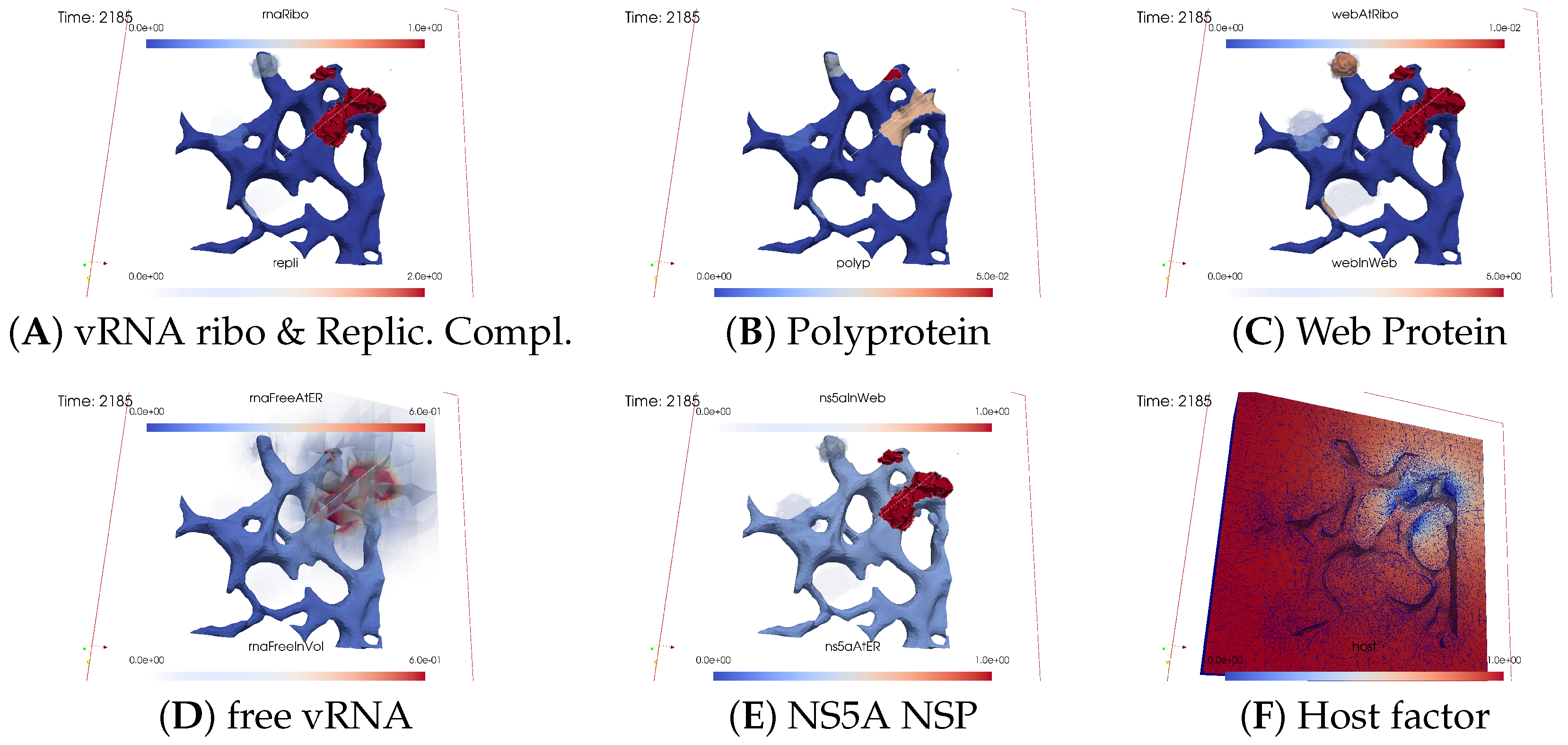
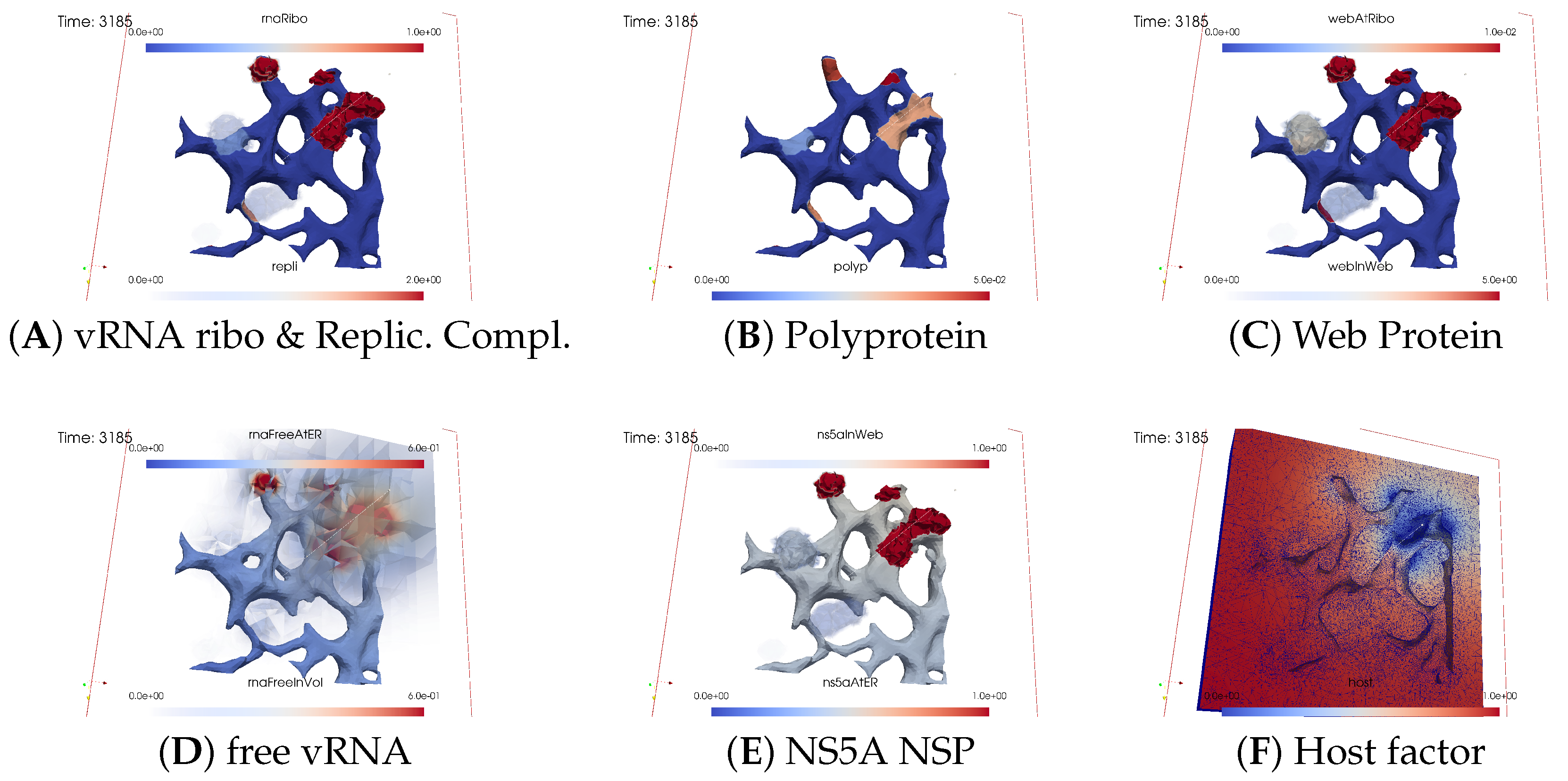
Ordering of All Simulation Screenshots
- A
- Ribosomal bound vRNA (“RNA ribo”) on the ribosomal zones of the ER surface and the replication complex (RC) in the membranous webs (MWs) volume.
- B
- Polyprotein on the ribosomal zones of the ER surface.
- C
- Web protein (WP) in the ribosomal zones of the ER surface and membranous web (MW) volumes.
- D
- Free viral RNA (vRNA) in the volume and at the ER surface.
- E
- NS5A at the ER surface and in the membranous web volumes.
- F
- Host factor overall in the volume.
Initial State: One vRNA Attached to One Ribosomal Zone
Viral Protein Production, Movement, and MW Zone Activation
vRNA Polymerization and Propagation
Closing of the vRNA Cycle
Illumination of MW Spots Like a Domino Effect
Remarks on Visualization Properties
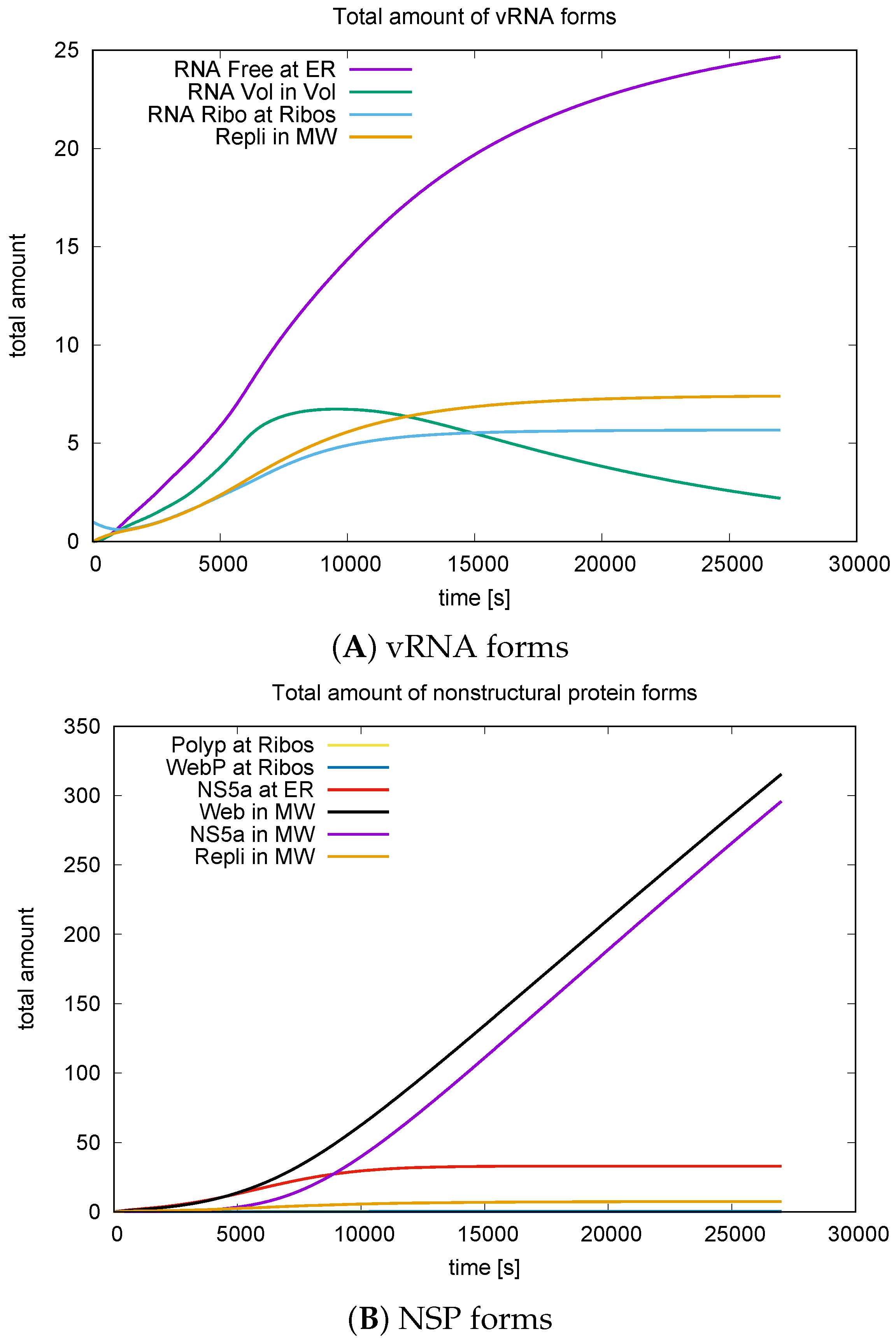
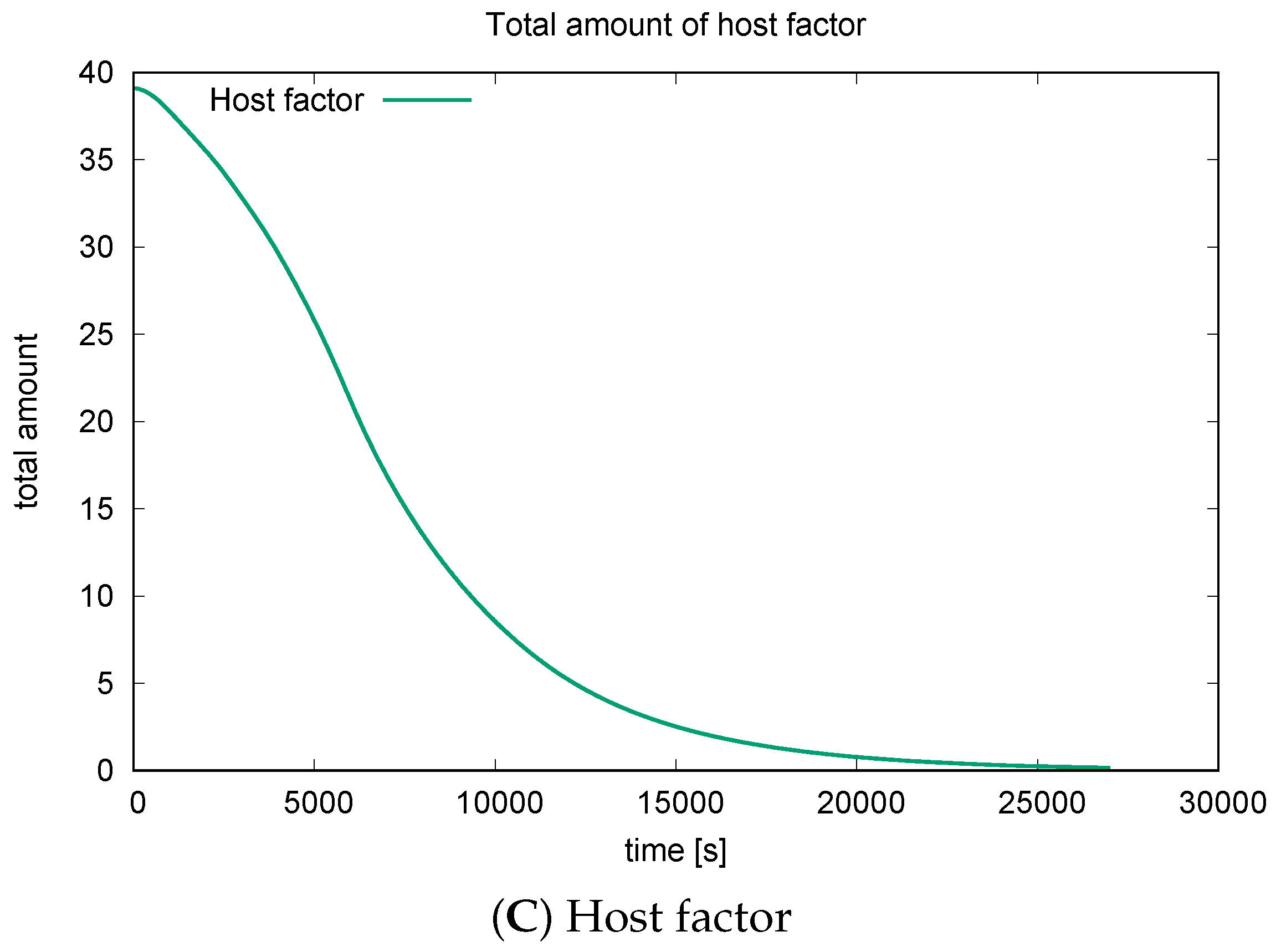
3.3. Quantitative Data Evaluations and Numerical Robustness
3.4. Mass Conservation along the Exchange between Manifold and Volume
4. Discussion
4.1. Model Components and Their Respective Spatial Dynamics
4.2. ER Surface Remodeling/MW Zone Establishment
4.3. vRNA Movement and Location
4.4. Replication Complex and the Cis Condition
4.5. Nonlinear Diffusion and Reaction Coefficient Structure
4.6. Comparison to Quantitative Experimental Data
4.7. Quantitative Model Validation—Spatial Patterns and Parameter Set
4.8. Relationship between Form and Function
4.9. Qualitative Model Properties
4.10. Aims of Our Work and Milestones
- Establish mathematical equations describing the biophysics of intracellular virus replication in a fully 3D spatio-temporal resolved manner.
- Develop a computational framework capable of efficiently and robustly simulating these equations, which comprises:
- (a)
- Interpolation of experimental spatio-temporal virus replication states
- (b)
- Extrapolation of experimental spatio-temporal virus replication states to times beyond those already measured.
- (c)
- Prediction of spatio-temporal patterns of interactions among major components of intracellular virus replication.
- The 3D in silico “computer simulation screenshots”/states complement observable 3D “in vitro microscope screenshots”/states.
- Ensure the framework’s flexibility for extensions and adaptions. Particularly, for future applications it should enable in silico probing of the action of direct antiviral agents (DAAs) on spatio-temporal virus replication dynamics.
4.11. Summary: In Silico Microscope
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PDE | partial differential equation |
| sufPDE | surface PDE |
| GMG | geometric multi grid |
| SLE | system of linear equations |
| ER | endoplasmic reticulum |
| MW | membranous web |
| FRAP | fluorescence recovery after photobleaching |
| vRNA | viral RNA |
| NSP | non structural protein |
| SP | structural protein |
| WP | Web Protein |
| NS5A | HCV NSP # 5A |
| NS5B | HCV NSP # 5B |
| RC | Replication Complex |
| FRAP | Fluorescence Recovery After Photobleaching |
Appendix A. Additional Details Concerning Technical Grid and Model Details
Appendix A.1. Extended Description of the Surface Reconstruction Procedure

Appendix A.2. Coupled sufPDE/PDE System, Other Representations
Appendix A.2.1. Abbreviated Version of Model Equations
Appendix A.2.2. Colored Version of Model Equations
Appendix A.3. Coupled Surface Volume Models in the Literature
Appendix B. Cis Requirement—Short Description
Appendix B.1. Biological Basis of Cis Requierement
Appendix B.2. Model Construction and Cis Requirement
References
- Bakrania, S.; Chavez, C.; Ipince, A.; Rocca, M.; Oliver, S.; Stansfield, C.; Subrahmanian, R. Impacts of Pandemics and Epidemics on Child Protection: Lessons learned from a rapid review in the context of COVID-19. In Innocenti Working Paper 2020-05; UNICEF Office of Research—Innocenti: Florence, Italy, 2020. [Google Scholar]
- WHO. COVID-19 Strategic Preparedness and Response Plan: Monitoring and Evaluation Framework; WHO/WHE/2021.07; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- WHO. 2021 Mid-Year Report: WHO Strategic Action against COVID-19; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- WHO. Scoping Review of Interventions to Maintain Essential Services for Maternal, Newborn, Child and Adolescent Health and Older People during Disruptive Events; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- WHO. Global Hepatitis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- WHO. WHO Strategic Response Plan: West Africa Ebola Outbreak. In WHO Library Cataloguing-in-Publication Data; World Health Organization: Geneva, Switzerland, 2015; ISBN 9789241508698. [Google Scholar]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus- strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Bühler, S.; Bartenschlager, R. Chronic viral hepatitis and its association with liver cancer. Biol. Chem. 2017, 398, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Bartenschlager, R. Dengue virus and Hepatitis C virus-induced replication and assembly compartments: The enemy inside - caught in the web. J. Virol. 2014, 88, 5907–5911. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Path. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed]
- Belda, O.; Targett-Adams, P. Small molecule inhibitors of the hepatitis C virus-encoded NS5A protein. Vir. Res. 2012, 170, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Targett-Adams, P.; Graham, E.; Middleton, J.; Palmer, A.; Shaw, S.; Lavender, H.; Brain, P.; Tran, T.; Jones, L.; Wakenhut, F.; et al. Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: Insights into compound modes of action. J. Virol. 2011, 85, 6353–6368. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Regal, R.E.; Mohammad, R.A. Daclatasvir: A NS5A Replication Complex Inhibitor for Hepatitis C Infection. Ann. Pharmacother. 2016, 50, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Guedj, J.; Rong, L.; Dahari, H.; Perelson, A. A perspective on modelling hepatitis C virus infection. J. Vir. Hep. 2010, 17, 825–833. [Google Scholar] [CrossRef]
- Rong, L.; Guedj, J.; Dahari, H.; Coffield, D.J.; Levi, M.; Smith, P.; Perelson, A. Analysis of hepatitis C virus decline during treatment with the protease inhibitor danoprevir using a multiscale model. PLoS Comp. Biol. 2013, 9, e1002959. [Google Scholar] [CrossRef]
- Dahari, H.; Guedj, J.; Perelson, A.; Layden, T. Hepatitis C Viral Kinetics in the Era of Direct Acting Antiviral Agents and IL28B. Curr. Hepatol. Rep. 2011, 10, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Churkin, A.; Lewkiewicz, S.; Reinharz, V.; Dahari, H.; Barash, D. Efficient Methods for Parameter Estimation of Ordinary and Partial Differential Equation Models of Viral Hepatitis Kinetics. Mathematics 2020, 8, 1483. [Google Scholar] [CrossRef] [PubMed]
- Dahari, H.; Ribeiro, R.; Rice, C.; Perelson, A. Mathematical Modeling of Subgenomic Hepatitis C Virus Replication in Huh-7 Cells. J. Virol. 2007, 81, 750–760. [Google Scholar] [CrossRef]
- Dahari, H.; Sainz, B.; Sainz, J.; Perelson, A.; Uprichard, S. Modeling Subgenomic Hepatitis C Virus RNA Kinetics during Treatment with Alpha Interferon. J. Virol. 2009, 83, 6383–6390. [Google Scholar] [CrossRef]
- Adiwijaya, B.; Herrmann, E.; Hare, B.; Kieffer, T.; Lin, C.; Kwong, A.D.; Garg, V.; Randle, J.C.R.; Sarrazin, C.; Zeuzem, S.; et al. A Multi-Variant, Viral Dynamic Model of Genotype 1 HCV to Assess the in vivo Evolution of Protease-Inhibitor Resistant Variants. PLoS Comp. Biol. 2010, 6, e1000745. [Google Scholar] [CrossRef]
- Binder, M.; Sulaimanov, N.; Clausznitzer, D.; Schulze, M.; Hüber, C.; Lenz, S.; Schlöder, J.; Trippler, M.; Bartenschlager, R.; Lohmann, V.; et al. Replication vesicles are load- and choke-points in the hepatitis C virus lifecycle. PLoS Path. 2013, 9, e1003561. [Google Scholar] [CrossRef]
- Hattaf, K.; Yousfi, N. Global stability for reaction-diffusion equations in biology. Comput. Math. Appl. 2013, 66, 1488–1497. [Google Scholar] [CrossRef]
- Hattaf, K.; Yousfi, N. A generalized HBV model with diffusion and two delays. Comput. Math. Appl. 2015, 69, 31–40. [Google Scholar] [CrossRef]
- Hattaf, K.; Yousfi, N. A numerical method for delayed partial differential equations describing infectious diseases. Comput. Math. Appl. 2016, 72, 2741–2750. [Google Scholar] [CrossRef]
- Wang, K.; Wang, W. Propagation of HBV with spatial dependence. Math. Biosci. 2007, 210, 78–95. [Google Scholar] [CrossRef]
- Xu, R.; Ma, Z. An HBV model with diffusion and time delay. J. Theoret. Biol. 2009, 257, 499–509. [Google Scholar] [CrossRef]
- Chi, N.C.; Avila Vales, E.; Garcia Almeida, G. Analysis of a HBV model with diffusion and time delay. J. Appl. Math. 2012, 25, 578561. [Google Scholar]
- Zhang, Y.; Xu, Z. Dynamics of a diffusive HBV model with delayed Beddington? DeAngelis response. Nonlinear Anal. RWA 2014, 15, 118–139. [Google Scholar] [CrossRef]
- Targett-Adams, P.; Boulant, S.; McLauchlan, J. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 2008, 82, 2182–2195. [Google Scholar] [CrossRef]
- Knodel, M.M.; Reiter, S.; Targett-Adams, P.; Grillo, A.; Herrmann, E.; Wittum, G. 3D Spatially Resolved Models of the Intracellular Dynamics of the Hepatitis C Genome Replication Cycle. Viruses 2017, 9, 282. [Google Scholar] [CrossRef]
- Knodel, M.M.; Reiter, S.; Targett-Adams, P.; Grillo, A.; Herrmann, E.; Wittum, G. Advanced Hepatitis C Virus Replication PDE Models within a Realistic Intracellular Geometric Environment. Int. J. Environ. Res. Public Health 2019, 16, 513. [Google Scholar] [CrossRef] [PubMed]
- Knodel, M.M.; Nägel, A.; Reiter, S.; Rupp, M.; Vogel, A.; Targett-Adams, P.; McLauchlan, J.; Herrmann, E.; Wittum, G. Quantitative analysis of Hepatitis C NS5A viral protein dynamics on the ER surface. Viruses 2018, 10, 28. [Google Scholar] [CrossRef]
- Knodel, M.M.; Wittum, G.; Vollmer, J. Efficient Estimates of Surface Diffusion Parameters for Spatio-Temporally Resolved Virus Replication Dynamics. Int. J. Mol. Sci. 2024, 25, 2993. [Google Scholar] [CrossRef] [PubMed]
- Knodel, M.M.; Nägel, A.; Herrmann, E.; Wittum, G. PDE Models of Virus Replication Coupling 2D Manifold and 3D Volume Effects Evaluated at Realistic Reconstructed Cell Geometries. In Finite Volumes for Complex Applications X Volume 1, Elliptic and Parabolic Problems; FVCA 2023; Franck, E., Fuhrmann, J., Michel-Dansac, V., Navoret, L., Eds.; Proceedings in Mathematics & Statistics; Springer: Cham, Switzerland, 2023; Volume 432. [Google Scholar] [CrossRef]
- Knodel, M.M.; Nägel, A.; Wittum, G. Solving nonlinear virus replication PDE models with hierarchical grid distribution based GMG. In Submitted to ENUMATH23 Proceedings; Springer: Berlin/Heidelberg, Germany, 2024. [Google Scholar]
- Kühnel, W. Differential Geometry: Curves–Surfaces–Manifolds; American Mathematical Society: Providence, RI, USA, 2005. [Google Scholar]
- Dziuk, G.; Elliott, C.M. Surface finite elements for parabolic equations. J. Comput. Math. 2007, 25, 385–407. [Google Scholar]
- Knabner, P.; Angermann, L. Numerical Methods for Elliptic and Parabolic Partial Differential Equations; Springer: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Nägel, A.; Schulz, V.; Siebenborn, M.; Wittum, G. Scalable shape optimization methods for structured inverse modeling in 3D diffusive processes. Comput. Vis. Sci. 2015, 17, 79–88. [Google Scholar] [CrossRef]
- Bank, R.E.; Rose, D. Some Error Estimates for the Box Method. SIAM J. Nu. Anal. 1987, 24, 777–787. [Google Scholar] [CrossRef]
- Eyre, N.; Fiches, G.; Aloia, A.; Helbig, K.; McCartney, E.; McErlean, C.; Li, K.; Aggarwal, A.; Turville, S.G.; Bearda, M. Dynamic imaging of the hepatitis C virus NS5A protein during a productive infection. J. Virol. 2014, 88, 3636–3652. [Google Scholar] [CrossRef] [PubMed]
- Chukkapalli, V.; Berger, K.L.; Kelly, S.M.; Thomas, M.; Deiters, A.; Randall, G. Daclatasvir inhibits hepatitis C virus NS5A motility and hyper-accumulation of phosphoinositides. Virology 2015, 476, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential Role of Domain III of Nonstructural Protein 5A for Hepatitis C Virus Infectious Particle Assembly. PLoS Path. 2010, 4, e1000035. [Google Scholar] [CrossRef] [PubMed]
- Hackbusch, W. On first and second order box schemes. Computing 1989, 41, 277–296. [Google Scholar] [CrossRef]
- Reiter, S.; Vogel, A.; Heppner, I.; Rupp, M.; Wittum, G. A massively parallel geometric multigrid solver on hierarchically distributed grids. Comp. Vis. Sci. 2013, 16, 151–164. [Google Scholar] [CrossRef]
- Einstein, A. Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen. Ann. Der Phys. 1905, 17, 549–560. [Google Scholar] [CrossRef]
- Knodel, M.M.; Nägel, A.; Reiter, S.; Rupp, M.; Vogel, A.; Targett-Adams, P.; Herrmann, E.; Wittum, G. Multigrid analysis of spatially resolved hepatitis C virus protein simulations. Comput. Vis. Sci. 2015, 17, 235–253. [Google Scholar] [CrossRef]
- Jones, D.; Gretton, S.; McLauchlan, J.; Targett-Adams, P. Mobility analysis of an NS5A-GFP fusion protein in cells actively replicating hepatitis C virus subgenomic RNA. J. Gen. Vir. 2007, 88, 470–475. [Google Scholar] [CrossRef]
- Fiches, G.N.; Eyre, N.S.; Aloia, A.L.; Van Der Hoek, K.; Betz-Stablein, B.; Luciani, F.; Chopra, A.; Beard, M.R. HCV RNA traffic and association with NS5A in living cells. Virology 2016, 493, 60–74. [Google Scholar] [CrossRef]
- Lohmann, V. Hepatitis C Virus RNA Replication. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Current Topics in Microbiology and Immunology; Bartenschlager, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 369. [Google Scholar]
- Appel, N.; Herian, U.; Bartenschlager, R. Efficient rescue of hepatitis C virus RNA replication by trans-complementation with nonstructural protein 5A. J. Virol. 2005, 79, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Randall, G. Spatiotemporal Analysis of Hepatitis C Virus Infection. PLoS Pathog. 2015, 11, e1004758. [Google Scholar] [CrossRef]
- Vainio, J.; Cutts, F. Yellow fever. In Global Programme for Vaccines and Immunization; World Health Organization: Geneva, Switzerland, 1998. [Google Scholar]
- Kling, K.; Domingo, C.; Bogdan, C.; Duffy, S.; Harder, T.; Howick, J. Duration of protection after vaccination against yellow fever—Systematic review and meta-analysis. Clin. Infect. Dis. 2022, 75, 2266–2274. [Google Scholar] [CrossRef] [PubMed]
- Hilversum, N.; Scientific Volume Imaging. Software. 2014. Available online: http://www.svi.nl/HuygensSoftware (accessed on 30 July 2017).
- van der Voort, H.; Brakenhoff, G. 3-D image formation in high-aperture fluorescence confocal microscopy: A numerical analysis. J. Microsc. 1990, 158, 43–54. [Google Scholar] [CrossRef]
- Kano, H.; van der Voort, H.; Schrader, M.; van Kempen, G.; Hell, S.W. Avalanche photodiode detection with object scanning and image restoration provides 2–4 fold resolution increase in two-photon fluorescence microscopy. Bioimaging 1996, 4, 87–197. [Google Scholar] [CrossRef]
- van Kempen, G.; van der Voort, H.; van Vliet, L. A quantitative comparison of two restoration methods as applied to confocal microscopy. J. Microsc. 1997, 185, 354–365. [Google Scholar] [CrossRef]
- Jungblut, D.; Queisser, G.; Wittum, G. Inertia Based Filtering of High Resolution Images Using a GPU Cluster. Comp. Vis. Sci. 2011, 14, 181–186. [Google Scholar] [CrossRef]
- Broser, P.; Schulte, R.; Roth, A.; Helmchen, F.; Waters, J.; Lang, S.; Sakmann, B.; Wittum, G. Nonlinear anisotropic diffusion filtering of three-dimensional image data from 2-photon microscopy. J. Biom. Opt. 2004, 9, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Reiter, S. Effiziente Algorithmen und Datenstrukturen für die Realisierung von Adaptiven, Hierarchischen Gittern auf Massiv Parallelen Systemen. Ph.D. Thesis, Goethe-Universität Frankfurt, Frankfurt am Main, Germany, 2015. [Google Scholar]
- Reiter, S.; Wittum, G. ProMesh Software; ProMesh4; ProMesh: Dossenheim, Germany, 2017; Available online: http://promesh3d.com/ (accessed on 19 April 2024).
- Reiter, S.; Logashenko, D.; Stichel, S.; Wittum, G.; Grillo, A. Models and simulations of variable-density flow in fractured porous media. Int. J. Comput. Sci. Eng. 2014, 9, 416–432. [Google Scholar] [CrossRef]
- Chernyshenko, A.Y.; Olshanskii, M.A.; Vassilevski, Y.V. A hybrid finite volume? Finite element method for bulk?surface coupled problems. J. Comput. Phys. 2018, 352, 516–533. [Google Scholar] [CrossRef]
- Bonetti, E.; Bonfanti, G.; Rossi, R. Analysis of a model coupling volume and surface processes in thermoviscoelasticity. Discret. Contin. Dyn. Syst. 2015, 35, 2349–2403. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
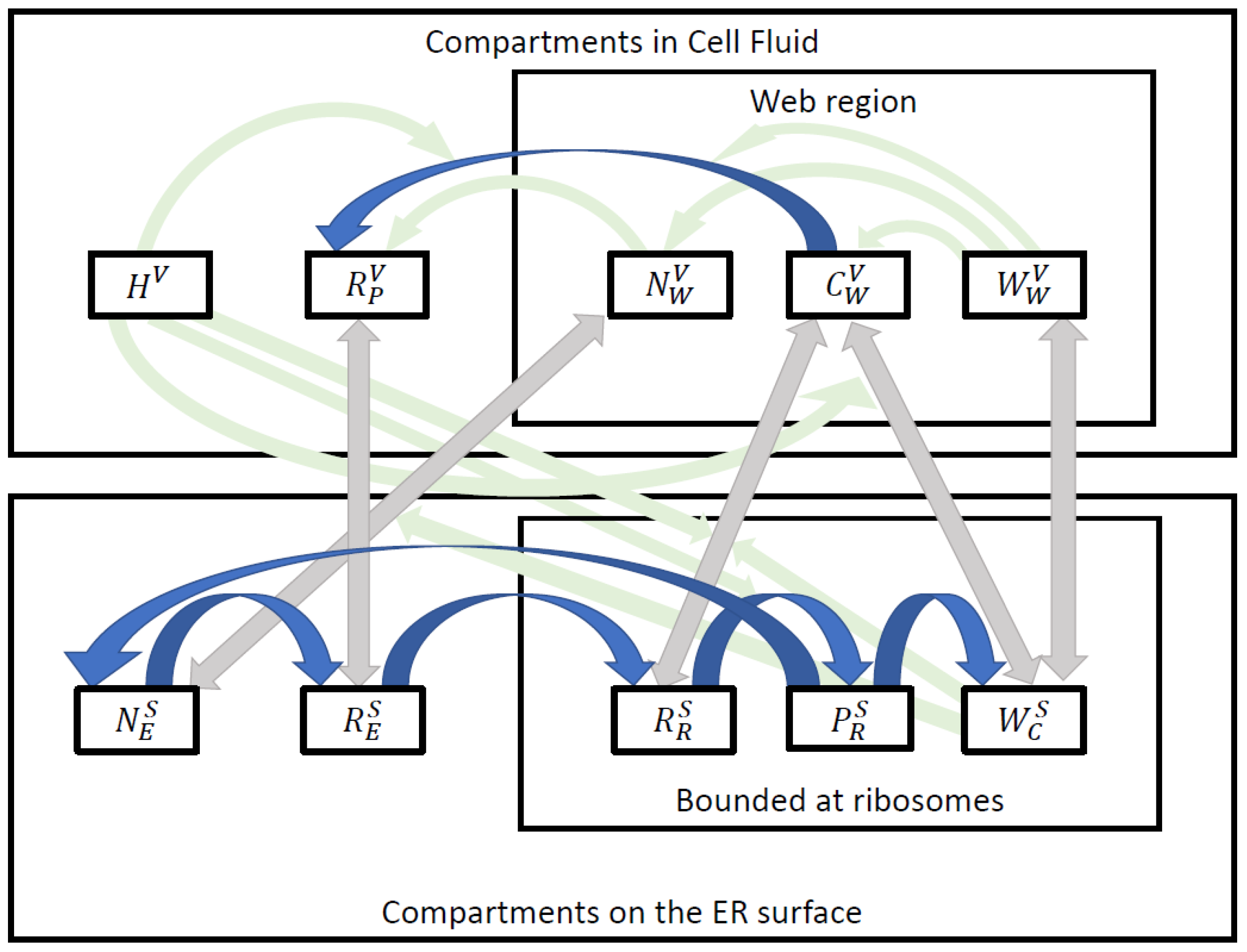
| Subdomain | Property |
|---|---|
| 2D manifold , embedded in 3D | |
| reconstructed ER surface except for | |
| 7 ribosomic zones: intersection ER/MW surfaces | |
| 2D closure of computational domain - hexahedron | |
| surface of enclosing hexahedron | |
| 3D volume | |
| cytosol (enclosed by the box enclosing in fact ) | |
| 7 MW subdomains: volume enclosed by MW surfaces | |
| Compartment | Region | Biophysical Interpretation |
|---|---|---|
| Concentration | ||
| Surface compartments (concentrations) defined at the 3D embedded curved 2D manifold | ||
| Ribosomal bound vRNA | ||
| Viral polyprotein translated at ribosomes | ||
| Web (NSP) protein/WP cleaved from the polyprotein | ||
| NS5A NSP cleaved from the polyprotein | ||
| polymerized free vRNA attached to the ER | ||
| Volume compartments (concentrations) defined in the 3D volume | ||
| Web (NSP) protein/WP detached from ribosomes to form MWs | ||
| NS5A NSP detached from ribosomes incorporated into MW | ||
| Replication complex/RC as combination of detached and | ||
| Polymerized free vRNA moving in the full volume | ||
| Host factor | ||
| Parameter | Value | Unit |
|---|---|---|
| m2 | ||
| 0.001 | ||
| 100./3600./10. = 0.0028 | ||
| 0.056 | ||
| 0.1 | ||
| 0.05 | ||
| 0.002 | ||
| 0.005 | ||
| 0.001 | ||
| 0.001 | ||
| 0.005 | ||
| 0.01 | ||
| 0.0005 | ||
| 0.0001 | ||
| 0.0005 | ||
| 0.01 | 1 | |
| 0.01 | 1 | |
| 1 | ||
| 0.01 | 1 | |
| 0.01 | 1 | |
| 0.01 | 1 | |
| 0.01 | 1 | |
| 0.01 | 1 | |
| 0.01 | 1 | |
| 0.01 | 1 | |
| 1 | 1 | |
| 1 , 0 else | 1 | |
| 1 , 0 else | 1 | |
| 1 | 1 | |
| 1 | 1 | |
| 1 | 1 | |
| b | 1 | |
| 1 |
| Level | DoFs | Vols |
|---|---|---|
| 0 | 96,370 | 41,446 |
| 1 | 667,280 | 331,568 |
| 2 | 4,890,410 | 2,652,544 |
| 3 | 37,266,790 | 21,220,352 |
| 4 | 290,579,070 | 169,762,816 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knodel, M.M.; Nägel, A.; Herrmann, E.; Wittum, G. Intracellular “In Silico Microscopes”—Comprehensive 3D Spatio-Temporal Virus Replication Model Simulations. Viruses 2024, 16, 840. https://doi.org/10.3390/v16060840
Knodel MM, Nägel A, Herrmann E, Wittum G. Intracellular “In Silico Microscopes”—Comprehensive 3D Spatio-Temporal Virus Replication Model Simulations. Viruses. 2024; 16(6):840. https://doi.org/10.3390/v16060840
Chicago/Turabian StyleKnodel, Markus M., Arne Nägel, Eva Herrmann, and Gabriel Wittum. 2024. "Intracellular “In Silico Microscopes”—Comprehensive 3D Spatio-Temporal Virus Replication Model Simulations" Viruses 16, no. 6: 840. https://doi.org/10.3390/v16060840
APA StyleKnodel, M. M., Nägel, A., Herrmann, E., & Wittum, G. (2024). Intracellular “In Silico Microscopes”—Comprehensive 3D Spatio-Temporal Virus Replication Model Simulations. Viruses, 16(6), 840. https://doi.org/10.3390/v16060840