
Reconstructing Prehistoric Viral Genomes from Neanderthal Sequencing Data

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples, Representative Sequences, and Reference Sequences
2.2. Genome Assembly by Reference Mapping
2.3. Tracking and Quantification of DNA Damage Patterns in DNA Sequence Reads
2.4. Viral Genealogies
2.5. Assemblies with Random Reference Sequences
3. Results
3.1. Taxonomic Assessment of Neanderthal Raw Genomic Data
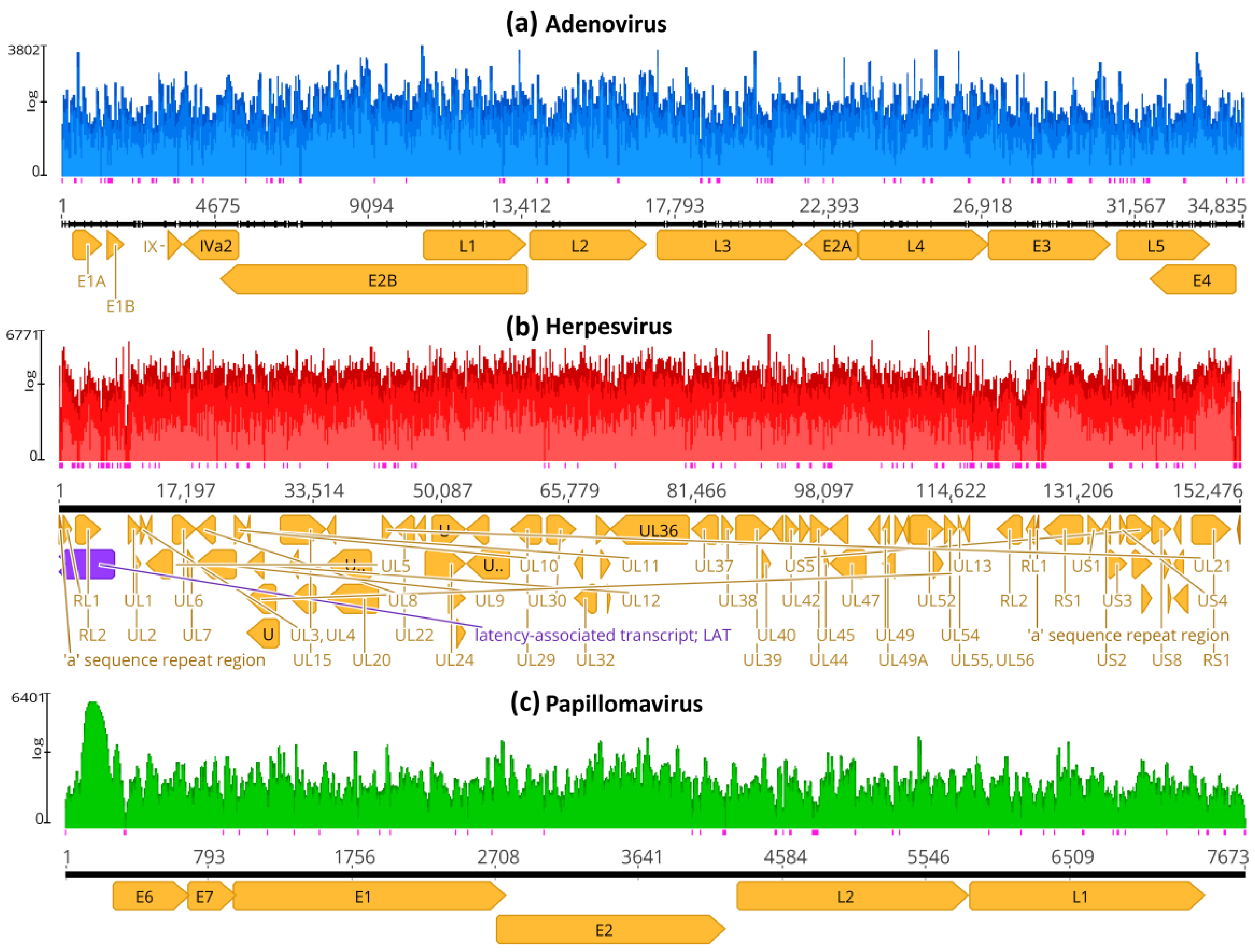
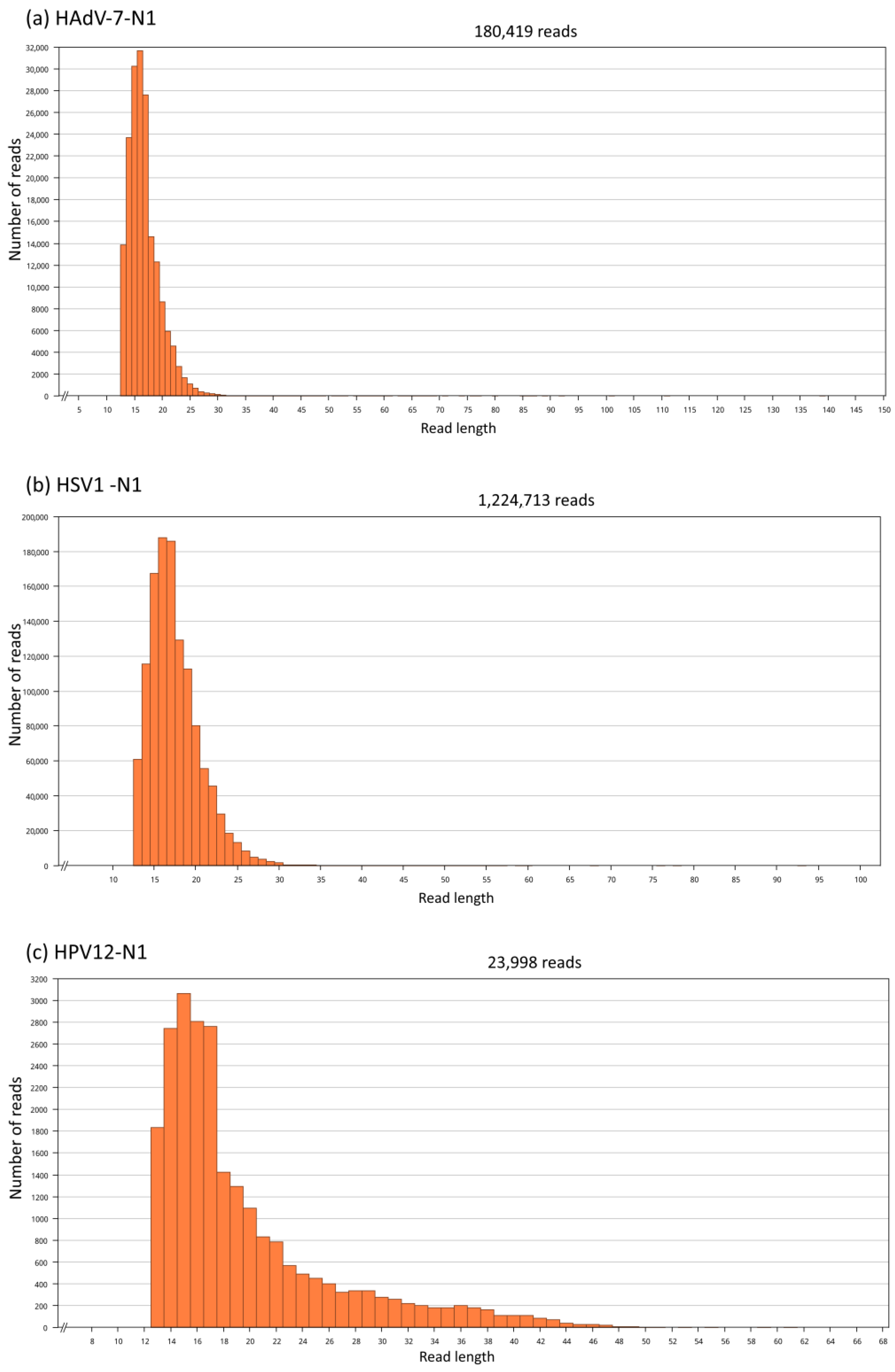
3.2. Genome Assembly by Mapping
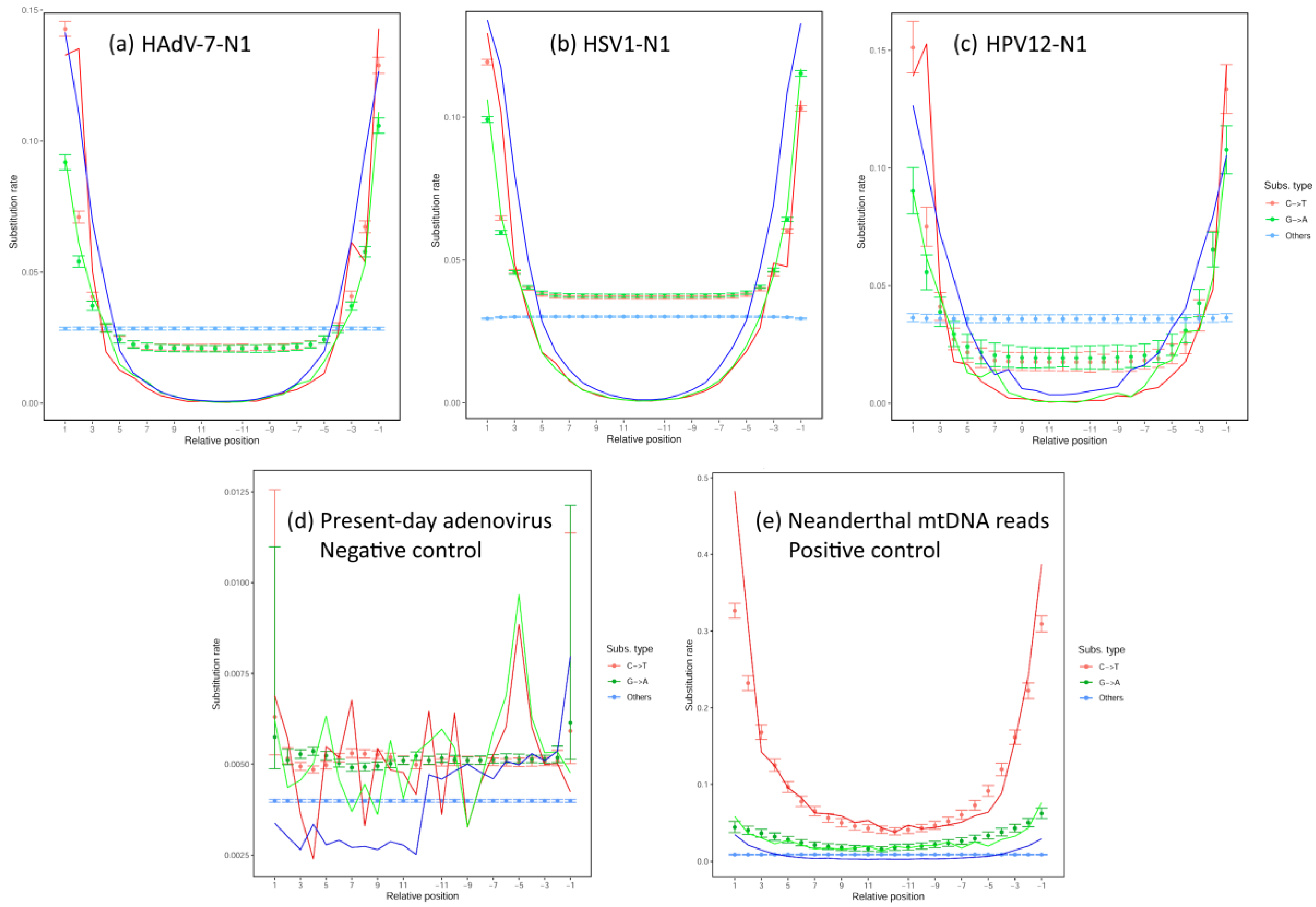
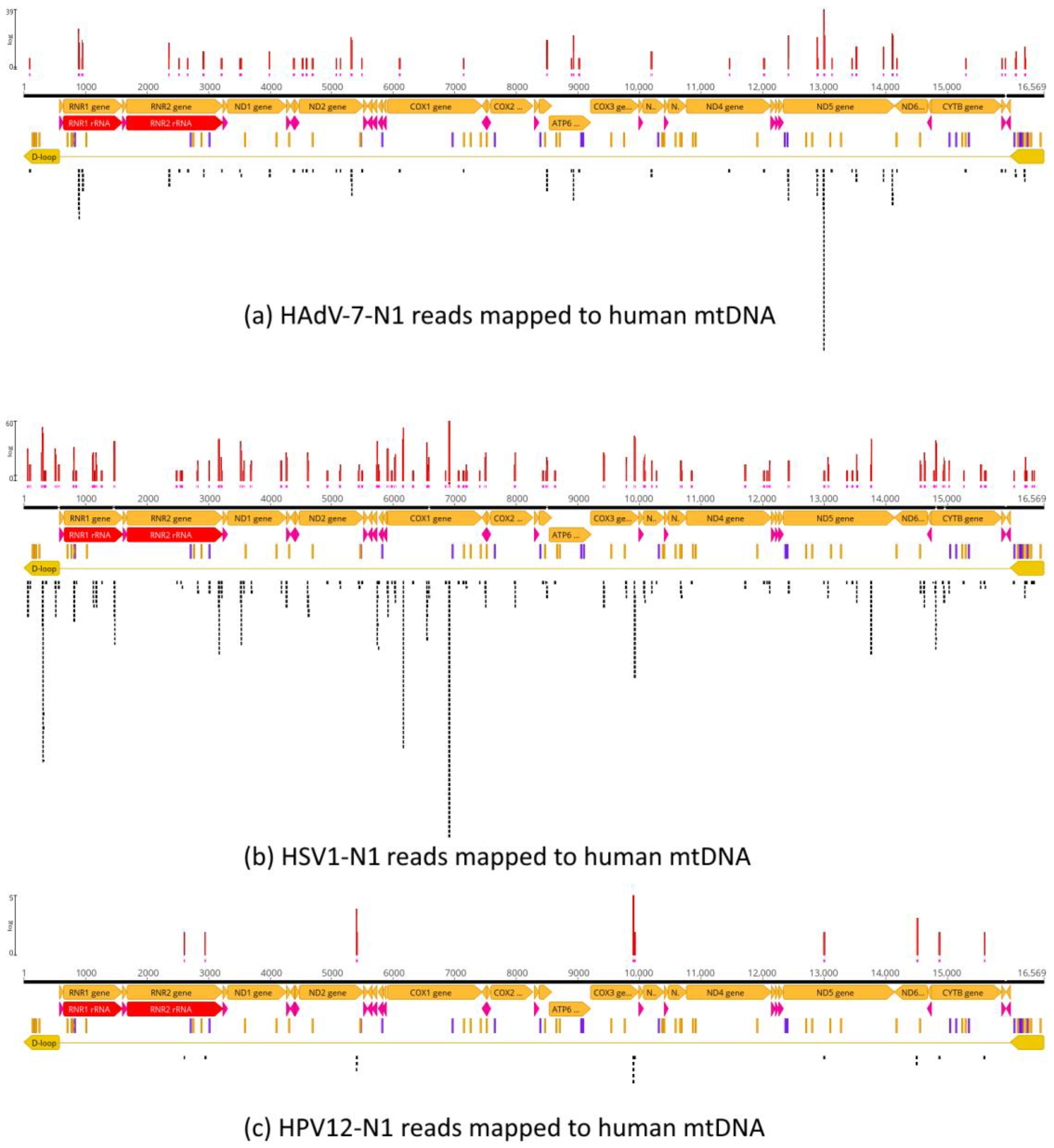
3.3. Analysis of Deamination Patterns in Neanderthal Genomic Reads
3.4. Genome Mapping with Taxonomic Reference Sequences
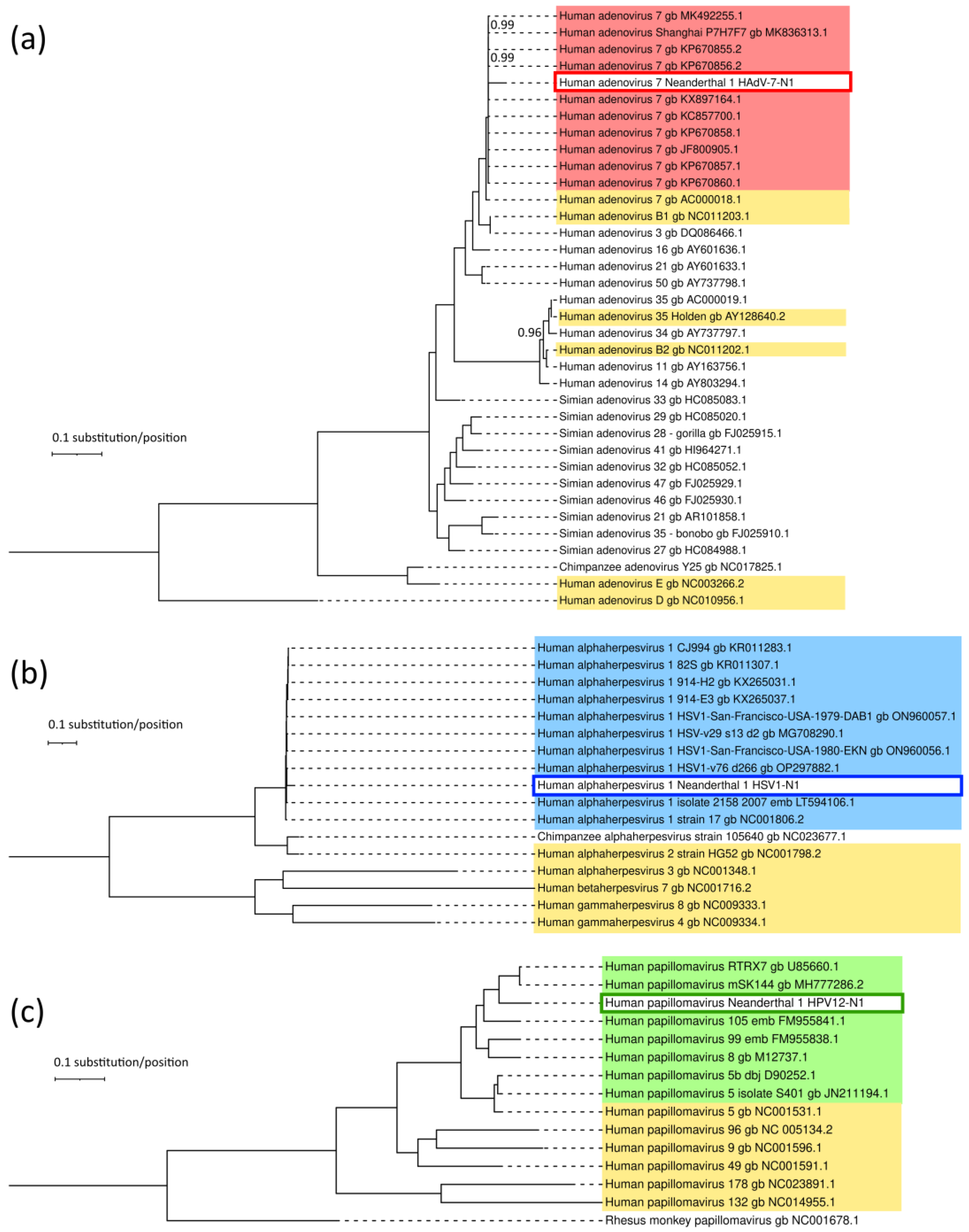
3.5. Genetic Relatedness of Neanderthal Viruses and Extant Viruses
3.6. Putative Changes in Proteins of Neanderthal Viruses
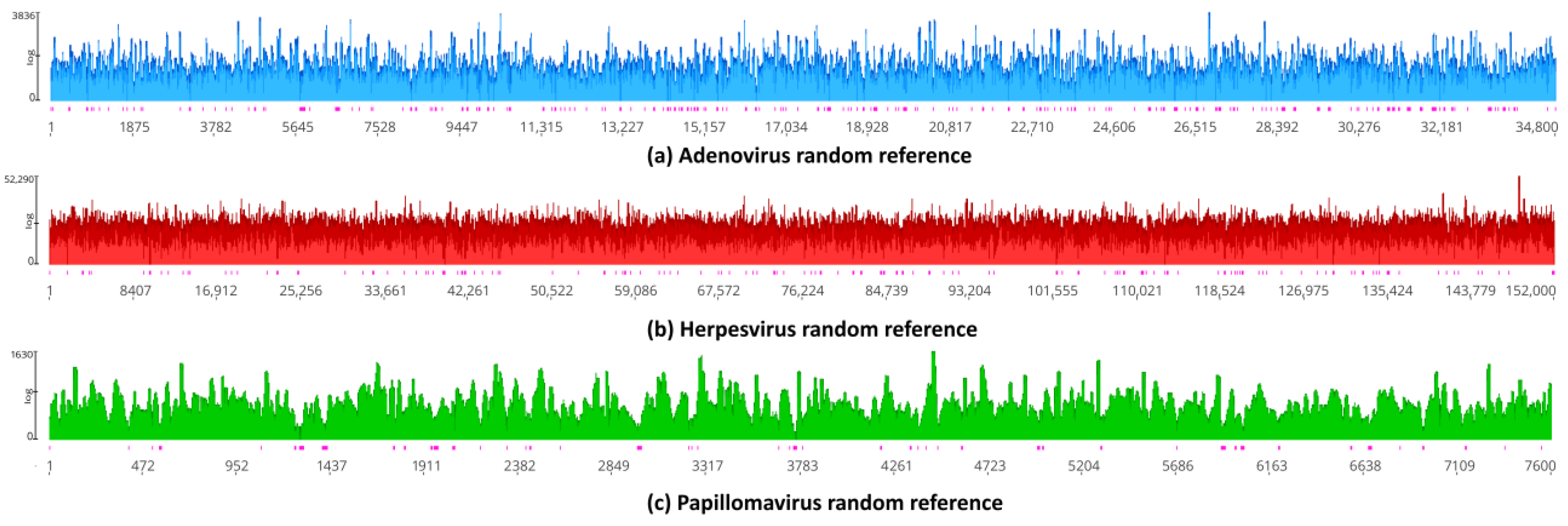
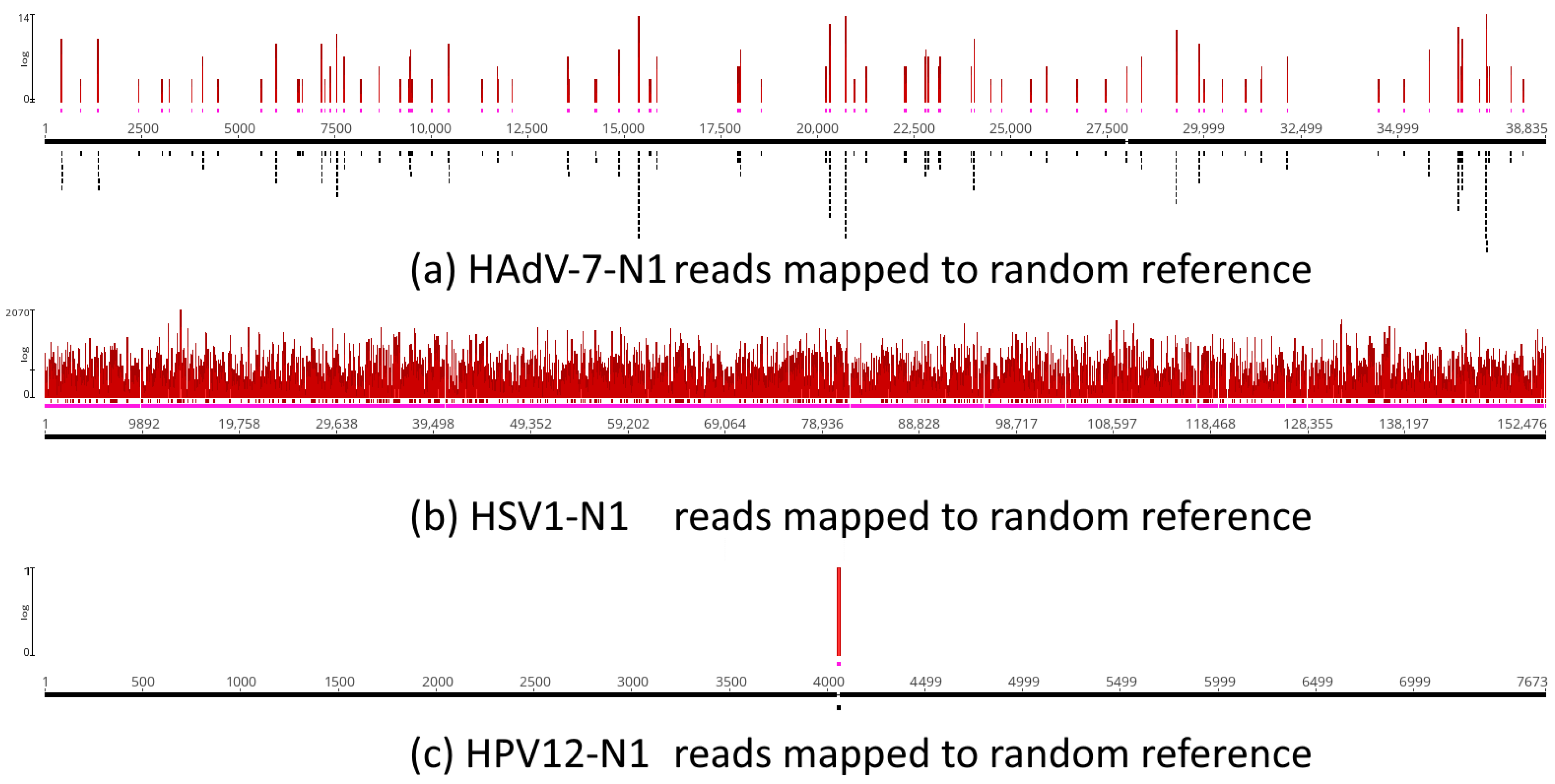
3.7. Assessment of Random Effects in Genome Mapping
3.8. Control with a Nonpersistent DNA Virus
3.9. Herpesvirus Type 1 (HSV1) and Type 2 (HSV2)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banks, W.E.; d’Errico, F.; Peterson, A.T.; Kageyama, M.; Sima, A.; Sánchez-Goñi, M.-F. Neanderthal Extinction by Competitive Exclusion. PLoS ONE 2008, 3, e3972. [Google Scholar] [CrossRef] [PubMed]
- Jones, D. The Neanderthal Within. New Sci. 2007, 193, 28–32. [Google Scholar] [CrossRef]
- Dalén, L.; Orlando, L.; Shapiro, B.; Brandström-Durling, M.; Quam, R.; Gilbert, M.T.P.; Díez Fernández-Lomana, J.C.; Willerslev, E.; Arsuaga, J.L.; Götherström, A. Partial Genetic Turnover in Neandertals: Continuity in the East and Population Replacement in the West. Mol. Biol. Evol. 2012, 29, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, G.; Getz, W.M.; Rosenberg, N.A.; Feldman, M.W.; Hovers, E.; Kolodny, O. Disease Transmission and Introgression Can Explain the Long-Lasting Contact Zone of Modern Humans and Neanderthals. Nat. Commun. 2019, 10, 5003. [Google Scholar] [CrossRef] [PubMed]
- Enard, D.; Petrov, D.A. Evidence That RNA Viruses Drove Adaptive Introgression between Neanderthals and Modern Humans. Cell 2018, 175, 360–371.e13. [Google Scholar] [CrossRef] [PubMed]
- Wolff, H.; Greenwood, A.D. Did Viral Disease of Humans Wipe out the Neandertals? Med. Hypotheses 2010, 75, 99–105. [Google Scholar] [CrossRef]
- Chayavichitsilp, P.; Buckwalter, J.V.; Krakowski, A.C.; Friedlander, S.F. Herpes Simplex. Pediatr. Rev. 2009, 30, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Guellil, M.; van Dorp, L.; Inskip, S.A.; Dittmar, J.M.; Saag, L.; Tambets, K.; Hui, R.; Rose, A.; D’Atanasio, E.; Kriiska, A.; et al. Ancient Herpes Simplex 1 Genomes Reveal Recent Viral Structure in Eurasia. Sci. Adv. 2022, 8, eabo4435. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. Human Papillomaviruses: Diversity, Infection and Host Interactions. Nat. Rev. Microbiol. 2022, 20, 95–108. [Google Scholar] [CrossRef]
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B. ICTV Virus Taxonomy Profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef]
- Robinson, C.M.; Singh, G.; Lee, J.Y.; Dehghan, S.; Rajaiya, J.; Liu, E.B.; Yousuf, M.A.; Betensky, R.A.; Jones, M.S.; Dyer, D.W.; et al. Molecular Evolution of Human Adenoviruses. Sci. Rep. 2013, 3, 1812. [Google Scholar] [CrossRef]
- Hassan, M.M.; Li, D.; El-Deeb, A.S.; Wolff, R.A.; Bondy, M.L.; Davila, M.; Abbruzzese, J.L. Association Between Hepatitis B Virus and Pancreatic Cancer. J. Clin. Oncol. 2008, 26, 4557–4562. [Google Scholar] [CrossRef] [PubMed]
- Speck, S.H.; Ganem, D. Viral Latency and Its Regulation: Lessons from the Gamma-Herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef]
- Sankararaman, S.; Mallick, S.; Dannemann, M.; Prüfer, K.; Kelso, J.; Pääbo, S.; Patterson, N.; Reich, D. The Genomic Landscape of Neanderthal Ancestry in Present-Day Humans. Nature 2014, 507, 354–357. [Google Scholar] [CrossRef]
- Spyrou, M.A.; Bos, K.I.; Herbig, A.; Krause, J. Ancient Pathogen Genomics as an Emerging Tool for Infectious Disease Research. Nat. Rev. Genet. 2019, 20, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Lion, T. Adenovirus Persistence, Reactivation, and Clinical Management. FEBS Lett. 2019, 593, 3571–3582. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Underdown, S.J. Neanderthal Genomics Suggests a Pleistocene Time Frame for the First Epidemiologic Transition. Am. J. Phys. Anthropol. 2016, 160, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Naqvi, A.; Bair, E.; Nares, S.; Khan, A.A. Viral MicroRNAs Identified in Human Dental Pulp. J. Endod. 2017, 43, 84–89. [Google Scholar] [CrossRef]
- Kazi, M.M.A.G.; Bharadwaj, R.; Bhat, K.; Happy, D. Association of Herpes Viruses with Mild, Moderate and Severe Chronic Periodontitis. J. Clin. Diagn. Res. 2015, 9, DC05–DC08. [Google Scholar] [CrossRef]
- Baringer, J.R.; Swoveland, P. Recovery of Herpes-Simplex Virus from Human Trigeminal Ganglions. N. Engl. J. Med. 1973, 288, 648–650. [Google Scholar] [CrossRef]
- Juhl, D.; Mosel, C.; Nawroth, F.; Funke, A.-M.; Dadgar, S.M.; Hagenström, H.; Kirchner, H.; Hennig, H. Detection of Herpes Simplex Virus DNA in Plasma of Patients with Primary but Not with Recurrent Infection: Implications for Transfusion Medicine? Transfus. Med. 2010, 20, 38–47. [Google Scholar] [CrossRef]
- Skov, L.; Peyrégne, S.; Popli, D.; Iasi, L.N.M.; Devièse, T.; Slon, V.; Zavala, E.I.; Hajdinjak, M.; Sümer, A.P.; Grote, S.; et al. Genetic Insights into the Social Organization of Neanderthals. Nature 2022, 610, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Katz, K.S.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.R.; O’Sullivan, C. STAT: A Fast, Scalable, MinHash-Based k-Mer Tool to Assess Sequence Read Archive next-Generation Sequence Submissions. Genome Biol. 2021, 22, 270. [Google Scholar] [CrossRef]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate Paired Shotgun Read Merging via Overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. mapDamage2.0: Fast Approximate Bayesian Estimates of Ancient DNA Damage Parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database Indexing for Production MegaBLAST Searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods) [All Versions]. Available online: https://paup.phylosolutions.com/ (accessed on 23 November 2023).
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Wit, E.; van den Heuvel, E.; Romeijn, J.-W. ‘All Models Are Wrong…’: An Introduction to Model Uncertainty. Stat. Neerl. 2012, 66, 217–236. [Google Scholar] [CrossRef]
- Miller, M.A.; Schwartz, T.; Pickett, B.E.; He, S.; Klem, E.B.; Scheuermann, R.H.; Passarotti, M.; Kaufman, S.; O’Leary, M.A. A RESTful API for Access to Phylogenetic Tools via the CIPRES Science Gateway. 2015. Available online: https://journals.sagepub.com/doi/10.4137/EBO.S21501 (accessed on 23 November 2023).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; the UGENE team. Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.L. The Generalization of ‘Student’s’ Problem When Several Different Population Varlances Are Involved. Biometrika 1947, 34, 28–35. [Google Scholar] [CrossRef]
- Kulanayake, S.; Tikoo, S.K. Adenovirus Core Proteins: Structure and Function. Viruses 2021, 13, 388. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The Structural Basis of Herpesvirus Entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef]
- Ault, K.A. Epidemiology and Natural History of Human Papillomavirus Infections in the Female Genital Tract. Infect. Dis. Obstet. Gynecol. 2006, 2006, e40470. [Google Scholar] [CrossRef] [PubMed]
- Heegaard, E.D.; Brown, K.E. Human Parvovirus B19. Clin. Microbiol. Rev. 2002, 15, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Vafaie, J.; Schwartz, R.A. Parvovirus B19 Infections. Int. J. Dermatol. 2004, 43, 747–749. [Google Scholar] [CrossRef]
- Lehmann, H.W.; von Landenberg, P.; Modrow, S. Parvovirus B19 Infection and Autoimmune Disease. Autoimmun. Rev. 2003, 2, 218–223. [Google Scholar] [CrossRef]
- Kaneko, H.; Kawana, T.; Ishioka, K.; Ohno, S.; Aoki, K.; Suzutani, T. Evaluation of Mixed Infection Cases with Both Herpes Simplex Virus Types 1 and 2. J. Med. Virol. 2008, 80, 883–887. [Google Scholar] [CrossRef]
- Susloparov, M.A.; Susloparov, I.M.; Zagoruĭko, T.I.; Noskova, N.V.; Makhova, N.M. Herpes simplex virus type 1 and 2 (HSV1,2) DNA detection by PCR during genital herpes. Mol. Genet. Mikrobiol. Virusol. 2006, 38–41. [Google Scholar]
- Farnsworth, A.; Wisner, T.W.; Webb, M.; Roller, R.; Cohen, G.; Eisenberg, R.; Johnson, D.C. Herpes Simplex Virus Glycoproteins gB and gH Function in Fusion between the Virion Envelope and the Outer Nuclear Membrane. Proc. Natl. Acad. Sci. USA 2007, 104, 10187–10192. [Google Scholar] [CrossRef] [PubMed]
- Kosulin, K.; Geiger, E.; Vécsei, A.; Huber, W.-D.; Rauch, M.; Brenner, E.; Wrba, F.; Hammer, K.; Innerhofer, A.; Pötschger, U.; et al. Persistence and Reactivation of Human Adenoviruses in the Gastrointestinal Tract. Clin. Microbiol. Infect. 2016, 22, e1–e381. [Google Scholar] [CrossRef] [PubMed]
- Gravitt, P.E. Evidence and Impact of Human Papillomavirus Latency. Open Virol. J. 2012, 6, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of Damage in Genomic DNA Sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wu, M.; Halpern, A.; Rusch, D.B.; Yooseph, S.; Frazier, M.; Venter, J.C.; Eisen, J.A. Stalking the Fourth Domain in Metagenomic Data: Searching for, Discovering, and Interpreting Novel, Deep Branches in Marker Gene Phylogenetic Trees. PLoS ONE 2011, 6, e18011. [Google Scholar] [CrossRef]
- Benkő, M.; Aoki, K.; Arnberg, N.; Davison, A.J.; Echavarría, M.; Hess, M.; Jones, M.S.; Kaján, G.L.; Kajon, A.E.; Mittal, S.K.; et al. ICTV Virus Taxonomy Profile: Adenoviridae. J. Gen. Virol. 2022, 103, 001721. [Google Scholar] [CrossRef] [PubMed]
- Gatherer, D.; Depledge, D.P.; Hartley, C.A.; Szpara, M.L.; Vaz, P.K.; Benkő, M.; Brandt, C.R.; Bryant, N.A.; Dastjerdi, A.; Doszpoly, A.; et al. ICTV Virus Taxonomy Profile: Herpesviridae. J. Gen. Virol. 2021, 102, 001673. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Chen, Z.; Bernard, H.-U.; Chan, P.K.S.; DeSalle, R.; Dillner, J.; Forslund, O.; Haga, T.; McBride, A.A.; Villa, L.L.; et al. ICTV Virus Taxonomy Profile: Papillomaviridae. J. Gen. Virol. 2018, 99, 989–990. [Google Scholar] [CrossRef]
- Xu, G.J.; Kula, T.; Xu, Q.; Li, M.Z.; Vernon, S.D.; Ndung’u, T.; Ruxrungtham, K.; Sanchez, J.; Brander, C.; Chung, R.T.; et al. Comprehensive Serological Profiling of Human Populations Using a Synthetic Human Virome. Science 2015, 348, aaa0698. [Google Scholar] [CrossRef]
- Burn, A.; Roy, F.; Freeman, M.; Coffin, J.M. Widespread Expression of the Ancient HERV-K (HML-2) Provirus Group in Normal Human Tissues. PLoS Biol. 2022, 20, e3001826. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Chagyrskaya | # Reads | DNA Viruses | |
|---|---|---|---|---|
| ERR10073004 | 01 | 5,187,065 | human gammaherpesvirus 4, human papillomavirus 12 and 36 | |
| ERR10073005 | 01 | 1,106,287 | human papillomavirus 20 | |
| ERR10073006 | 01 | 12,197,881 | human gammaherpesvirus 4, hepatitis B virus, human papillomavirus 10, 12, 19 and 209 | |
| ERR10073007 | 01 | 5,384,224 | Gammapapillomavirus 12 | |
| ERR10073008 | 01 | 581,197 | Not detected | |
| ERR10073009 | 01 | 5,434,074 | human alphaherpesvirus 1 | |
| ERR10073010 | 01 | 11,535,641 | human gammaherpesvirus 4, human papillomavirus 36 and human adenovirus 7 | |
| ERR10073011 | 01 | 2,145,096 | Not detected | |
| ERR10073012 | 01 | 813,154 | Not detected | |
| ERR10073013 | 01 | 5,836,042 | Papillomaviridae | |
| ERR10073014 | 01 | 10,283,890 | human papillomavirus 12 | |
| ERR10073060 | 07 | 31,299,145 | human papillomavirus 18, human respirovirus 1 and human mastadenovirus C | |
| ERR10073061 | 07 | 30,544,456 | human mastadenovirus C and human papillomavirus 16 | |
| ERR10073062 | 07 | 31,517,112 | human mastadenovirus C | |
| ERR10073063 | 07 | 34,004,864 | human alphaherpesvirus 2, Betaherpesvirinae Stealth virus 1 and human mastadenovirus C | |
| ERR10073064 | 07 | 34,089,383 | human mastadenovirus C and human papillomavirus 16 | |
| ERR10073065 | 07 | 34,548,780 | human betaherpesvirus 7 and Cytomegalovirus 1, Mastadenovirus | |
| ERR10073066 | 07 | 34,530,209 | human mastadenovirus C and human papillomavirus 16 | |
| ERR10073067 | 07 | 34,336,757 | human alphaherpesvirus 2 and Mastadenovirus | |
| ERR10073068 | 07 | 34,800,717 | Mastadenovirus | |
| ERR10073069 | 07 | 35,065,248 | human mastadenovirus C |
| RefSeq Accession | # Mapped Reads | GenBank ID | Virus | |
|---|---|---|---|---|
| Adenovirus | ||||
| AC_000018.1 | 78,255 | GI:56160876 | human adenovirus 7 | |
| NC_011203.1 | 54,835 | GI:197944726 | human adenovirus B1 | |
| NC_011202.1 | 8854 | GI:197944766 | human adenovirus B2 | |
| AC_000019.1 | 8573 | GI:56160914 | human adenovirus 35 | |
| NC_003266.2 | 6449 | GI:51527264 | human adenovirus E | |
| NC_010956.1 | 2716 | GI:190340974 | human adenovirus D | |
| Herpesvirus | ||||
| NC_001806.2 | 937,258 | GI:820945227 | human herpesvirus 1 17 | |
| NC_001798.2 | 167,080 | GI:820945149 | human herpesvirus 2 HG52 | |
| NC_009334.1 | 13,928 | GI:139424470 | human herpesvirus 4 | |
| NC_009333.1 | 10,443 | GI:139472801 | human herpesvirus 8 GK18 | |
| NC_001348.1 | 4056 | GI:9625875 | human herpesvirus 3 | |
| NC_001716.2 | 1857 | GI:51874225 | human herpesvirus 7 | |
| Papillomavirus | ||||
| NC_001531.1 | 1889 | GI:9627145 | human papillomavirus 5 | |
| NC_001591.1 | 494 | GI:9627363 | human papillomavirus 49 | |
| NC_005134.2 | 167 | GI:50253426 | human papillomavirus 96 | |
| NC_023891.1 | 82 | GI:607064610 | human papillomavirus 178 | |
| NC_001596.1 | 77 | GI:9627396 | human papillomavirus 9 | |
| NC_014955.1 | 76 | GI:319962668 | human papillomavirus 132 |
| Real | Random | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Coverage | Mean | SD | # Reads | Mean | SD | # Reads | Welch’s t | c.v. | p-Value |
| HAdV-7-N1 | 102.2 | 249.4 | 180,419 | 62.2 | 180.2 | 126,613 | 51.58 | 1.64 | 0 |
| HSV1-N1 | 171.2 | 247.4 | 1,224,713 | 154.8 | 704.3 | 1,166,326 | 23.78 | 1.64 | 0 |
| HPV12-N1 | 115.4 | 609.1 | 23,998 | 51.0 | 116.0 | 22,682 | 16.07 | 1.64 | 0 |
| Reference | Coverage Mean | Coverage SD | # Reads | Welch’s t | c.v. | p-Value |
|---|---|---|---|---|---|---|
| Parvovirus B19 | 2.0 | 9.2 | 714 | - | - | - |
| Random Ref. 1 | 3.5 | 16.9 | 1271 | −2.56 | 1.64 | 0.005267 |
| Random Ref. 2 | 7.2 | 41.6 | 2595 | 2.25 | 1.64 | 0.012046 |
| Random Ref. 3 | 2.7 | 10.0 | 975 | −1.48 | 1.64 | 0.068387 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, R.C.; Alves, G.V.; Ramon, M.; Antoneli, F.; Briones, M.R.S. Reconstructing Prehistoric Viral Genomes from Neanderthal Sequencing Data. Viruses 2024, 16, 856. https://doi.org/10.3390/v16060856
Ferreira RC, Alves GV, Ramon M, Antoneli F, Briones MRS. Reconstructing Prehistoric Viral Genomes from Neanderthal Sequencing Data. Viruses. 2024; 16(6):856. https://doi.org/10.3390/v16060856
Chicago/Turabian StyleFerreira, Renata C., Gustavo V. Alves, Marcello Ramon, Fernando Antoneli, and Marcelo R. S. Briones. 2024. "Reconstructing Prehistoric Viral Genomes from Neanderthal Sequencing Data" Viruses 16, no. 6: 856. https://doi.org/10.3390/v16060856
APA StyleFerreira, R. C., Alves, G. V., Ramon, M., Antoneli, F., & Briones, M. R. S. (2024). Reconstructing Prehistoric Viral Genomes from Neanderthal Sequencing Data. Viruses, 16(6), 856. https://doi.org/10.3390/v16060856