
Evolutionary and Phylogenetic Dynamics of SARS-CoV-2 Variants: A Genetic Comparative Study of Taiyuan and Wuhan Cities of China


Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Acquisition
2.2. Genome Annotation
Lineage and Subtyping Analysis
2.3. Sequence Alignment
2.4. Phylogenetic Tree Construction
2.5. Time-Based Phylogenetic Tree Construction
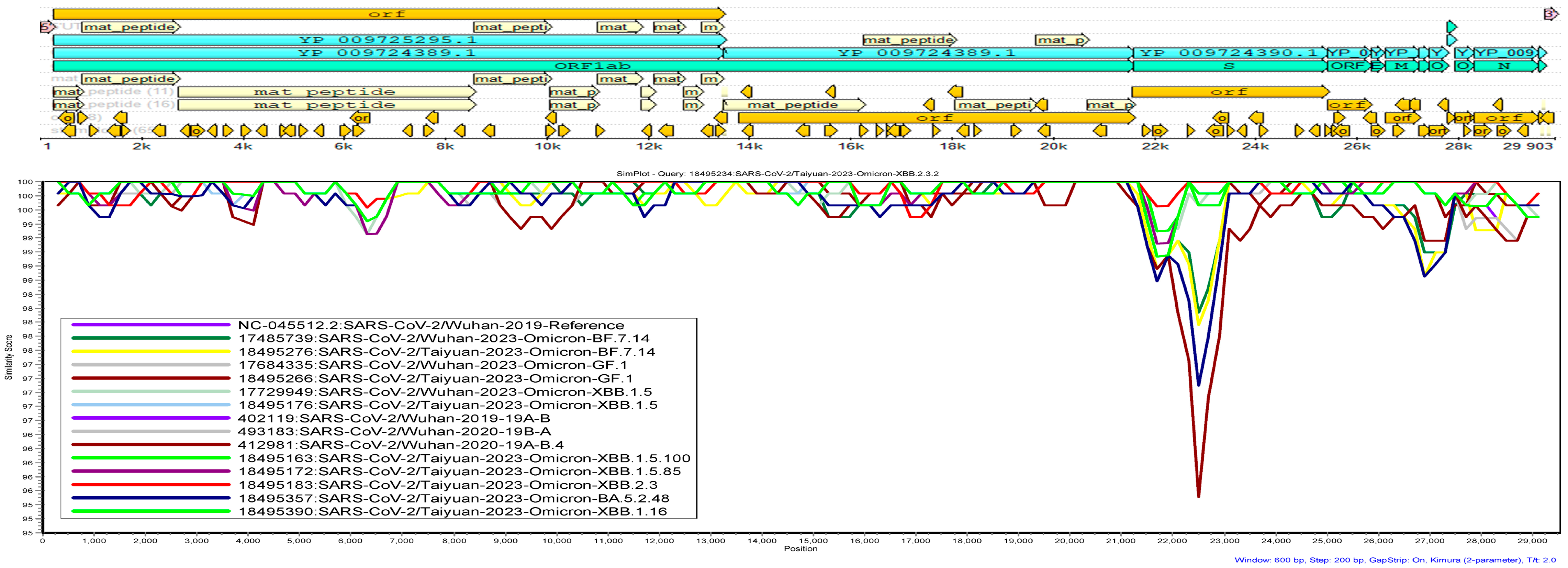
2.6. Genetic Similarity Analysis
2.7. Recombination Analysis
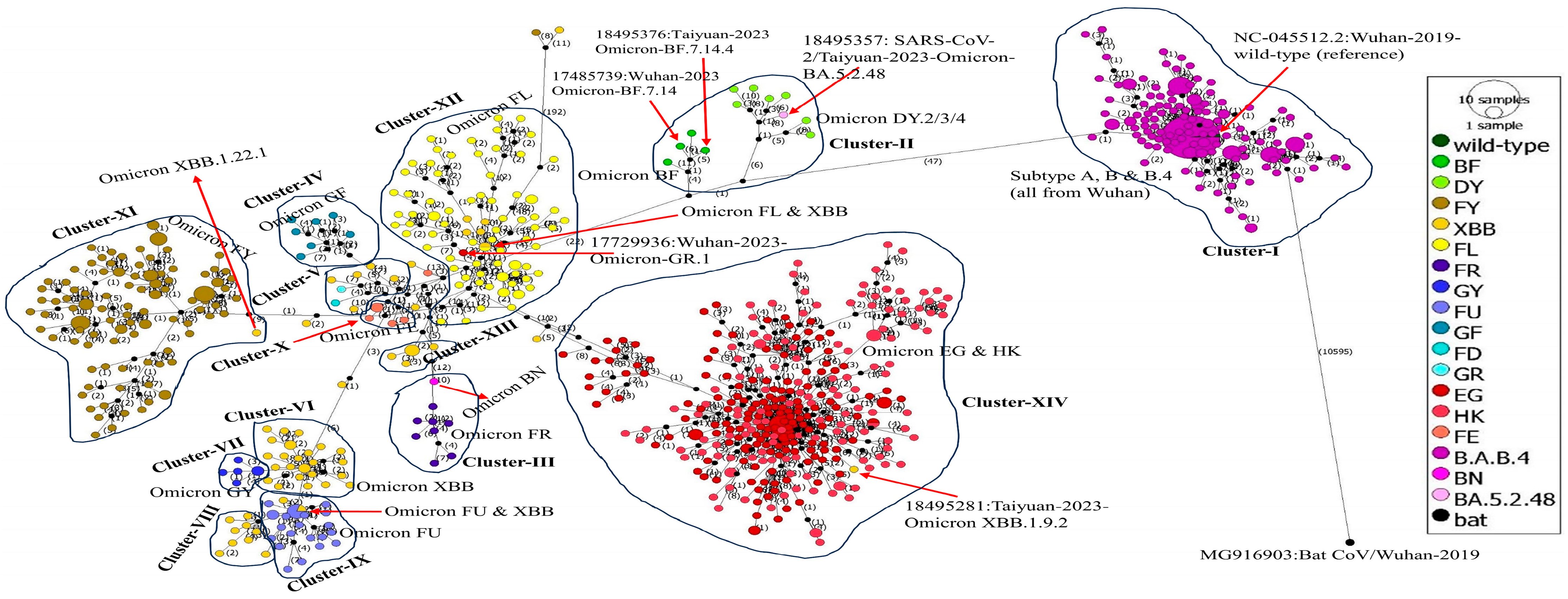
2.8. Phylogenetic Network Analysis
3. Results
3.1. Sequence Acquisition
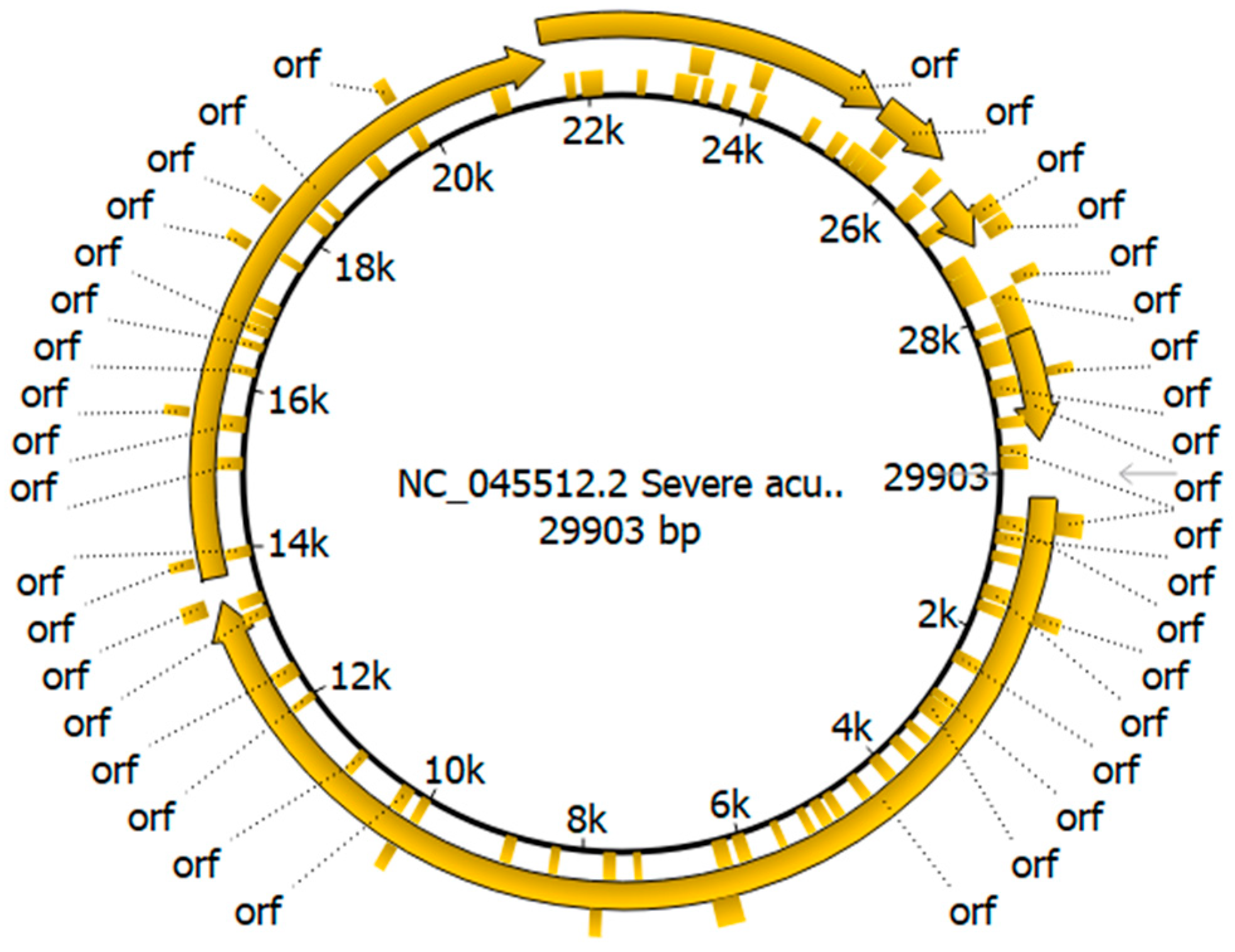
3.2. Genome Annotation
3.3. Subtype and Lineage Analysis
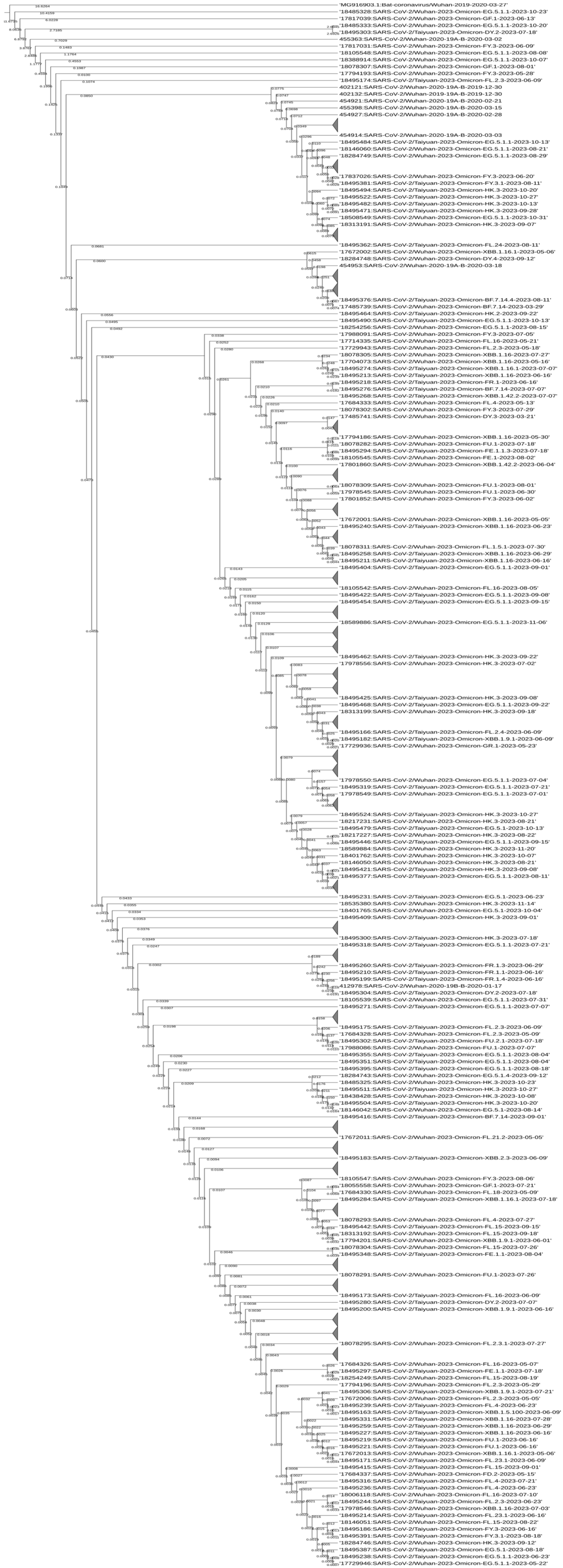
3.4. Phylogenetic Tree
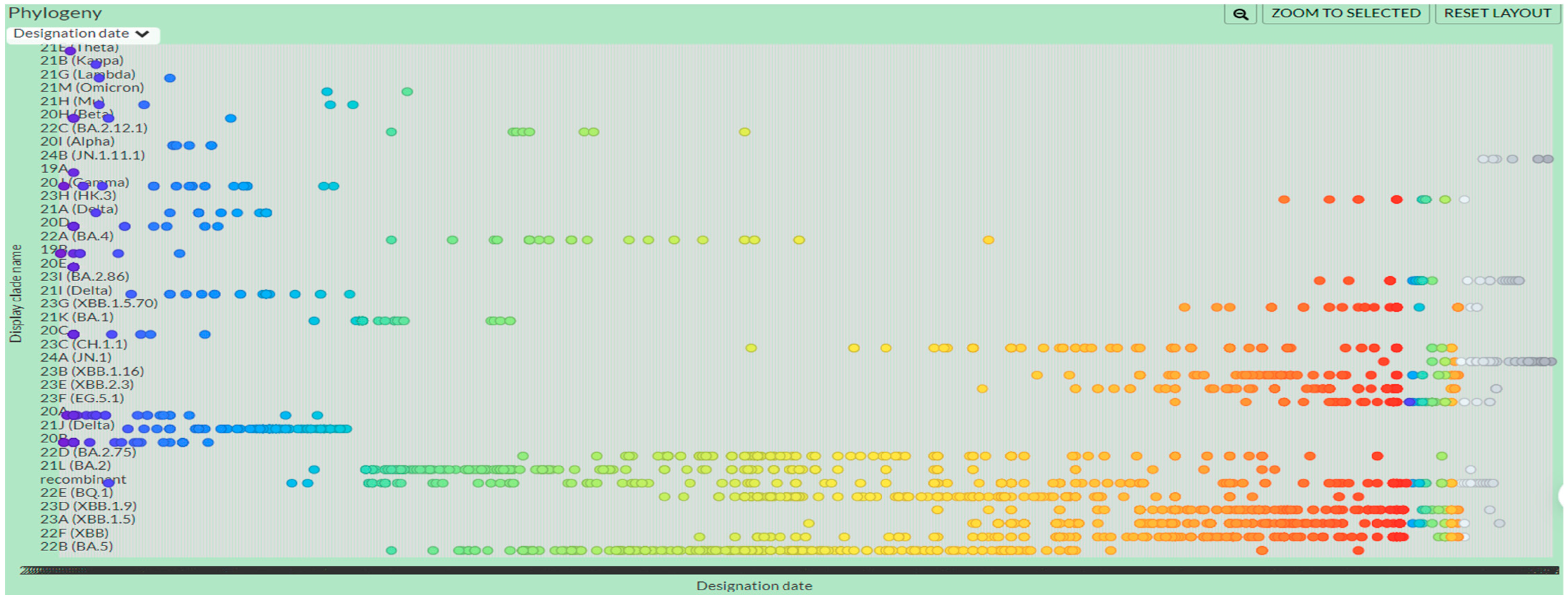
3.5. Time-Based Phylogenetic Tree
3.6. Genetic Similarity Analysis
3.7. Recombination Analysis
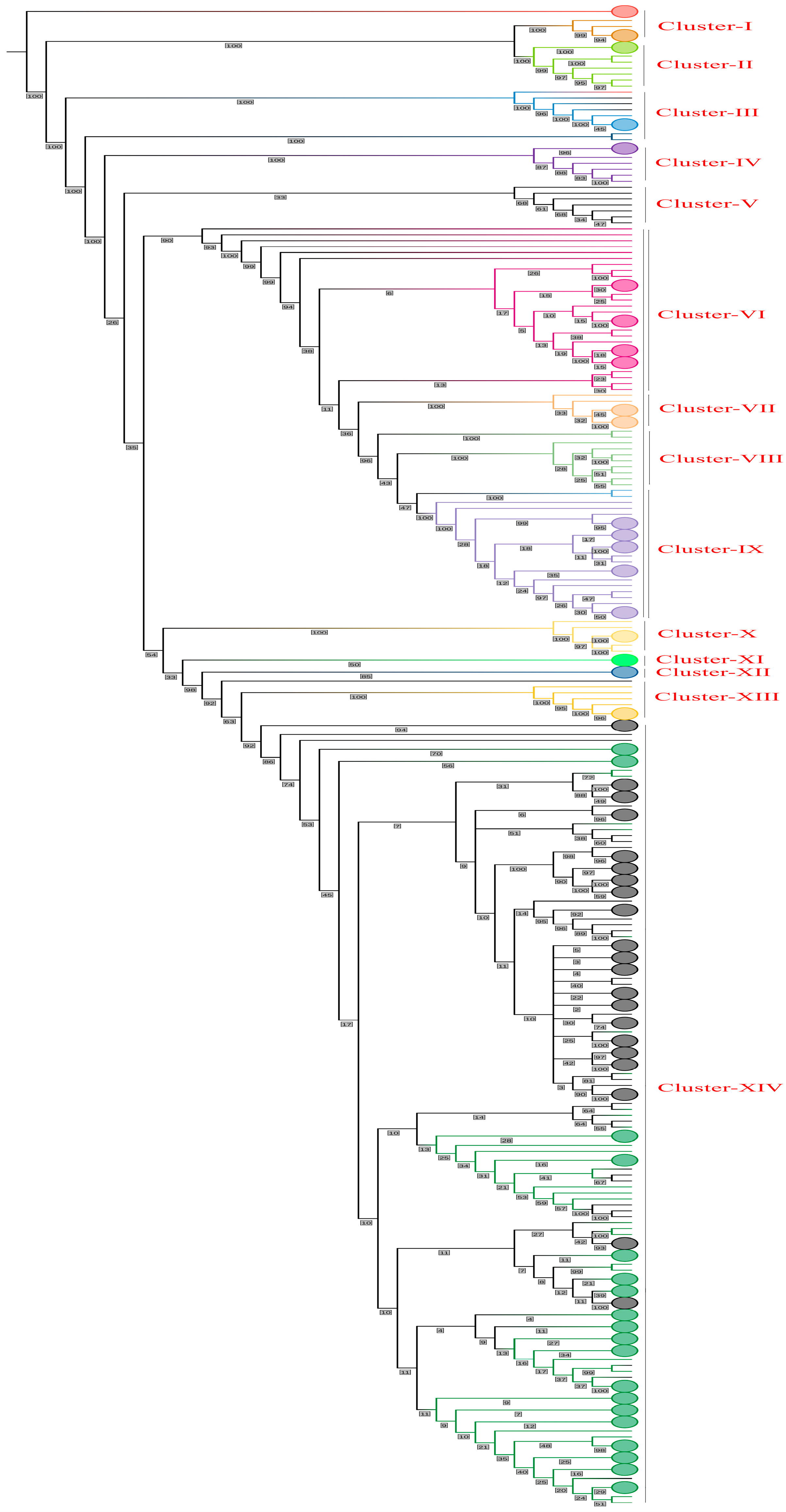
3.8. Phylogenetic Network Analysis
4. Discussion
4.1. Subtype and Lineage Analysis
4.2. Phylogenetic Analysis
4.3. Time-Based Phylogenetic Analysis
4.4. Genetic Similarity Analysis
4.5. Recombination Analysis
4.6. Phylogenetic Network Analysis
4.7. Importance of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Zhu, J.; Guo, J.; Xu, Y.; Chen, X. Viral dynamics of SARS-CoV-2 in saliva from infected patients. J. Infect. 2020, 81, e48–e50. [Google Scholar] [CrossRef]
- Cucinotta, D.; Vanelli, M. WHO declares COVID-19 a pandemic. Acta Biomedica Atenei Parmensis 2020, 91, 157. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Day, T.; Gandon, S.; Lion, S.; Otto, S.P. On the evolutionary epidemiology of SARS-CoV-2. Curr. Biol. 2020, 30, R849–R857. [Google Scholar] [CrossRef]
- Tang, D.; Comish, P.; Kang, R. The hallmarks of COVID-19 disease. PLoS Pathog. 2020, 16, e1008536. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e819. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Channabasappa, N.K.; Niranjan, A.K.; Emran, T.B. SARS-CoV-2 variant omicron XBB. 1.5: Challenges and prospects–correspondence. Int. J. Surg. 2023, 109, 1054–1055. [Google Scholar] [CrossRef]
- Haddad, D.; John, S.E.; Mohammad, A.; Hammad, M.M.; Hebbar, P.; Channanath, A.; Nizam, R.; Al-Qabandi, S.; Al Madhoun, A.; Alshukry, A. SARS-CoV-2: Possible recombination and emergence of potentially more virulent strains. PLoS ONE 2021, 16, e0251368. [Google Scholar] [CrossRef]
- Hussain, I.; Pervaiz, N.; Khan, A.; Saleem, S.; Shireen, H.; Wei, D.-Q.; Labrie, V.; Bao, Y.; Abbasi, A.A. Evolutionary and structural analysis of SARS-CoV-2 specific evasion of host immunity. Genes Immun. 2020, 21, 409–419. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Bhattacharjee, M.J.; Bhattacharya, A.; Kashyap, B.; Taw, M.J.; Li, W.-H.; Mukherjee, A.K.; Khan, M.R. Genome analysis of SARS-CoV-2 isolates from a population reveals the rapid selective sweep of a haplotype carrying many pre-existing and new mutations. Virol J. 2023, 20, 201. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Rehman, S.U.; Shafique, L.; Ihsan, A.; Liu, Q. Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. Pathogens 2020, 9, 240. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Team, U. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Etherington, G.J.; Dicks, J.; Roberts, I.N. Recombination Analysis Tool (RAT): A program for the high-throughput detection of recombination. Bioinformatics 2005, 21, 278–281. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2021. [Google Scholar]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef]
- Fengjiao, S.; Xiaodong, L.; Jian, L.; Hui, L. Epidemiologic characteristics of SARS-CoV-2 in Wuhan, other regions of China, and globally based on data gathered from January 2020 to February 2021. Medicine 2022, 101, e30019. [Google Scholar] [CrossRef]
- Sun, G.-Q.; Wang, S.-F.; Li, M.-T.; Li, L.; Zhang, J.; Zhang, W.; Jin, Z.; Feng, G.-L. Transmission dynamics of COVID-19 in Wuhan, China: Effects of lockdown and medical resources. Nonlinear Dyn. 2020, 101, 1981–1993. [Google Scholar] [CrossRef]
- Ma, Q.; Gao, J.; Zhang, W.; Wang, L.; Li, M.; Shi, J.; Zhai, Y.; Sun, D.; Wang, L.; Chen, B. Spatio-temporal distribution characteristics of COVID-19 in China: A city-level modeling study. BMC Infect. Dis. 2021, 21, 816. [Google Scholar] [CrossRef]
- Cicaloni, V.; Costanti, F.; Pasqui, A.; Niccolai, N.; Bongini, P. A bioinformatics approach to investigate structural and non-structural proteins in human coronaviruses. Front. Genet. 2022, 13, 891418. [Google Scholar] [CrossRef]
- Ahmadi, E.; Zabihi, M.R.; Hosseinzadeh, R.; Mohamed Khosroshahi, L.; Noorbakhsh, F. SARS-CoV-2 spike protein displays sequence similarities with paramyxovirus surface proteins; a bioinformatics study. PLoS ONE 2021, 16, e0260360. [Google Scholar] [CrossRef]
- Olivero, N.B.; Gonzalez-Reiche, A.S.; Re, V.E.; Castro, G.M.; Pisano, M.B.; Sicilia, P.; Barbas, M.G.; Khan, Z.; van de Guchte, A.; Dutta, J. Phylogenetic analysis and comparative genomics of SARS-CoV-2 from survivor and non-survivor COVID-19 patients in Cordoba, Argentina. BMC Genom. 2022, 23, 510. [Google Scholar] [CrossRef]
- Nie, Q.; Li, X.; Chen, W.; Liu, D.; Chen, Y.; Li, H.; Li, D.; Tian, M.; Tan, W.; Zai, J. Phylogenetic and phylodynamic analyses of SARS-CoV-2. Virus Res. 2020, 287, 198098. [Google Scholar] [CrossRef]
- Turkahia, Y.; Thornlow, B.; Hinrichs, A.; McBroome, J.; Ayala, N.; Ye, C.; De Maio, N.; Haussler, D.; Lanfear, R.; Corbett-Detig, R. Pandemic-scale phylogenomics reveals elevated recombination rates in the SARS-CoV-2 spike region. BioRxiv 2021. [Google Scholar] [CrossRef]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188.e5178. [Google Scholar] [CrossRef]
- Bolze, A.; Basler, T.; White, S.; Dei Rossi, A.; Wyman, D.; Dai, H.; Roychoudhury, P.; Greninger, A.L.; Hayashibara, K.; Beatty, M. Evidence for SARS-CoV-2 Delta and Omicron co-infections and recombination. Med 2022, 3, 848–859.e844. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses 2022, 14, 1239. [Google Scholar] [CrossRef]
- Al-Jawabreh, A.; Ereqat, S.; Dumaidi, K.; Al-Jawabreh, H.; Nasereddin, A. Complete genome sequencing of SARS-CoV-2 strains: A pilot survey in Palestine reveals spike mutation H245N. BMC Res. Notes 2021, 14, 466. [Google Scholar] [CrossRef]
- Tang, X.; Ying, R.; Yao, X.; Li, G.; Wu, C.; Tang, Y.; Li, Z.; Kuang, B.; Wu, F.; Chi, C. Evolutionary analysis and lineage designation of SARS-CoV-2 genomes. Sci. Bull. 2021, 66, 2297–2311. [Google Scholar] [CrossRef]
- Bai, Y.; Jiang, D.; Lon, J.R.; Chen, X.; Hu, M.; Lin, S.; Chen, Z.; Wang, X.; Meng, Y.; Du, H. Comprehensive evolution and molecular characteristics of a large number of SARS-CoV-2 genomes reveal its epidemic trends. Int. J. Infect. Dis. 2020, 100, 164–173. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Variant | City Name * | Total No. of Sequences |
|---|---|---|
| BF.7.14 | T (2), W (1) | 3 |
| DY.3 | W (2) | 2 |
| FY.3 | T (28), W (69) | 97 |
| XBB.1.16.1 | T (16), W (11) | 27 |
| XBB.1.16.1.1 | T (7), W (3) | 10 |
| FL.2.3 | T (7), W (14) | 21 |
| FL.13.1 | W (1) | 1 |
| FR.1.1 | T (4), W (1) | 5 |
| GY.1 | T (3), W (3) | 6 |
| FL.15 | T (2), W (7) | 9 |
| FL.21.2 | W (1) | 1 |
| FU.1 | T (9), W (13) | 22 |
| XBB.1.9.1 | T (3), W (2) | 5 |
| XBB.1.16.18 | W (2) | 2 |
| FL.16 | T (3), W (4) | 7 |
| FL.18 | W (1) | 1 |
| FL.4 | T (9), W (4) | 13 |
| GF.1 | T (2), W (5) | 7 |
| FD.2 | W (1) | 1 |
| FL.2.4 | T (4), W (2) | 6 |
| GR.1 | W (1) | 1 |
| EG.5.1.1 | T (113), W (44) | 157 |
| XBB.1.5 | T (1), W (2) | 3 |
| HK.5 | W (1) | 1 |
| FL.2 | T (3), W (3) | 6 |
| FL.21 | W (1) | 1 |
| EG.5.1 | T (9), W (5) | 14 |
| FY.3.1 | T (12), W (9) | 21 |
| XBB.1.42.2 | T (4), W (1) | 5 |
| DY.4 | W (2) | 2 |
| HK.1 | T (1), W (1) | 2 |
| HK.2 | T (4), W (1) | 5 |
| FU.2.1 | T (1), W (1) | 2 |
| XBB.1.42 | W (1) | 1 |
| HK.3 | T (68), W (58) | 126 |
| FL.2.3.1 | W (2) | 2 |
| FL.13.2 | W (1) | 1 |
| FL.1.5.1 | W (1) | 1 |
| FE.1 | W (1) | 1 |
| FE.1.1 | T (3), W (1) | 4 |
| HK.4 | W (4) | 4 |
| EG.5.1.4 | T (2), W (1) | 3 |
| 19A-B | W (167) | 167 |
| 19B-B | W (6) | 6 |
| 19B-A | W (20) | 20 |
| 19A-B.4 | W (3) | 3 |
| XBB.1.5.100 | T (1) | 1 |
| FL.23.1 | T (3) | 3 |
| XBB.1.5.85 | T (1) | 1 |
| XBB.2.3 | T (1) | 1 |
| FR.1.4 | T (1) | 1 |
| XBB.1.17.1 | T (1) | 1 |
| FR.1 | T (1) | 1 |
| BN.1.3.5 | T (1) | 1 |
| FL.4.6 | T (1) | 1 |
| XBB.1.9.2 | T (2) | 2 |
| FL.5 | T (2) | 2 |
| XBB.2.3.2 | T (1) | 1 |
| XBB.1.22.1 | T (1) | 1 |
| FR.1.3 | T (1) | 1 |
| FL.24 | T (3) | 3 |
| DY.2 | T (3) | 3 |
| FE.1.1.3 | T (1) | 1 |
| BA.5.2.48 | T (1) | 1 |
| BF.7.14.4 | T (1) | 1 |
| Event | Recombinant | Minor Parent | Major Parent | Detection * (RGBMCST) |
|---|---|---|---|---|
| 1 | 17672021:SARS-CoV-2/Wuhan-2023-Omicron-FY.3 | Unknown (455406:SARS-CoV-2/Wuhan-2020-19B-A) | 17684330:SARS-CoV-2/Wuhan-2023-Omicron-FL.18 | −−−−−−− |
| 2 | 17684330:SARS-CoV-2/Wuhan-2023-Omicron-FL.18 | 17729935:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.16 | 17672023:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.16 | −−−−−−− |
| 3 | 17672023:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.16 | 455406:SARS-CoV-2/Wuhan-2020-19B-A | Unknown (17672007:SARS-CoV-2/Wuhan-2023-Omicron-FL.13.1) | −+−++++ |
| 4 | 17684330:SARS-CoV-2/Wuhan-2023-Omicron-FL.18 | Unknown (455406:SARS-CoV-2/Wuhan-2020-19B-A) | 17672007:SARS-CoV-2/Wuhan-2023-Omicron-FL.13.1 | -−−−−−−+ |
| 5 | 17672021:SARS-CoV-2/Wuhan-2023-Omicron-FY.3 | 454997:SARS-CoV-2/Wuhan-2020-19A-B | Unknown (17978543:SARS-CoV-2/Wuhan-2023-Omicron-FY.3) | −++++++ |
| 6 | 17801857:SARS-CoV-2/Wuhan-2023-Omicron-FL.2.4 | Unknown (17672023:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.16) | 17672007:SARS-CoV-2/Wuhan-2023-Omicron-FL.13.1 | −+−−−−− |
| 7 | 17801857:SARS-CoV-2/Wuhan-2023-Omicron-FL.2.4 | 18284748:SARS-CoV-2/Wuhan-2023-Omicron-DY.4 | Unknown (17684337:SARS-CoV-2/Wuhan-2023-Omicron-FD.2) | −+−++++ |
| 8 | 18495231:SARS-CoV-2/Taiyuan-2023-Omicron-EG.5.1 | 18495199:SARS-CoV-2/Taiyuan-2023-Omicron-FR.1.4 | 18495364:SARS-CoV-2/Taiyuan-2023-Omicron-EG.5.1.1 | −−−−−−+ |
| 9 | 17801857:SARS-CoV-2/Wuhan-2023-Omicron-FL.2.4 | 455406:SARS-CoV-2/Wuhan-2020-19B-A | Unknown (17729949:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.5) | −−−−−−+ |
| Sr. No. | Network Method | Nucleotide Diversity | Segregating Sites | PI Sites * | Tajima’s D Statistic |
|---|---|---|---|---|---|
| 1 | TCS network | pi = 0.0142049 | 11466 | 1000 | D = −2.33398; p (D ≥ −2.33398) = 0.999236 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, B.; Wu, C. Evolutionary and Phylogenetic Dynamics of SARS-CoV-2 Variants: A Genetic Comparative Study of Taiyuan and Wuhan Cities of China. Viruses 2024, 16, 907. https://doi.org/10.3390/v16060907
Hussain B, Wu C. Evolutionary and Phylogenetic Dynamics of SARS-CoV-2 Variants: A Genetic Comparative Study of Taiyuan and Wuhan Cities of China. Viruses. 2024; 16(6):907. https://doi.org/10.3390/v16060907
Chicago/Turabian StyleHussain, Behzad, and Changxin Wu. 2024. "Evolutionary and Phylogenetic Dynamics of SARS-CoV-2 Variants: A Genetic Comparative Study of Taiyuan and Wuhan Cities of China" Viruses 16, no. 6: 907. https://doi.org/10.3390/v16060907
APA StyleHussain, B., & Wu, C. (2024). Evolutionary and Phylogenetic Dynamics of SARS-CoV-2 Variants: A Genetic Comparative Study of Taiyuan and Wuhan Cities of China. Viruses, 16(6), 907. https://doi.org/10.3390/v16060907