

Intrinsic Disorder in the Host Proteins Entrapped in Rabies Virus Particles


Abstract
:1. Introduction
- -
- Attachment/adsorption: At first, glycoprotein G of the virus interacts with the specific cell surface receptors [11];
- -
- -
- -
- -
- As the viral genome is tightly encapsidated by the viral nucleoprotein N, phosphoprotein P, and large protein L (or RNA-dependent RNA polymerase (RdRp)), upon its release into the cytoplasm, this RNP acts as the template for the transcription and replication processes catalyzed by the L-P polymerase complex [15];
- -
- Negri body (inclusion body or viral factory) formation: An RABV infection induces the formation of cytoplasmic inclusion bodies (Negri bodies [16]), the biogenesis of which is driven by liquid–liquid phase separation [17,18], which serve as viral factories, i.e., functional structures, where viral transcription and replication take place [15];
- -
- Transcription (or primary transcription): Since the genome of the RABV represents a linear, single-negative-stranded RNA, a viral-encoded RdRp (L protein) transcribes the viral antigenome RNA to mRNA in the cytoplasm [6,11]. Transcription leads to the synthesis of a positive-stranded leader RNA and five monocistronic capped and polyadenylated mRNAs;
- -
- Translation: A viral mRNA strand is used for the translation of five major proteins (N, P, M, G, and L);
- -
- Replication: RdRp replicates the progeny genome through a complementary replicative intermediate, the antigenome [6,11]. Here, “the RABV RdRp ignores the signals for mRNA synthesis on the genome to copy it into the positive-strand antigenome” [19]. After its antigenome is assembled into the RNP complex via its association with N, this replicative intermediate antigenome acts as a template for further rounds of replication to generate genomic RNA for progeny virions (antigenome is always always encapsidated by the N protein). Replication requires the newly synthesized N, P, and L proteins and a set of host factors;
- -
- Secondary transcription: New rounds of transcription (secondary transcription), translation, and replication take place following primary replication;
- -
- Assembly: All these viral particles (genome and proteins) assemble into new virions [11];
- -
- Budding: Assembled virions bud off from the cell surfaces of host cells, acquiring their envelope from the host cell membrane [20];
- -
- Release: The mature rabies virus normally releases from the cells through cell lysis and spreads through the central nervous system and brain to infect healthy cells [20].
2. Materials and Methods
2.1. Protein Datasets
2.2. Exploration of the Intrinsic Disorder Predisposition
2.3. ELMs: Eukaryotic Linear Motifs
2.4. Functional Annotation Derived from Disorder
2.5. FuzDrop Analysis: Identifying LLPS Promoters
2.6. Protein–Protein Interaction Network
2.7. CH-CDF Analysis
2.8. 3D Structures of Proteins
3. Results and Discussion
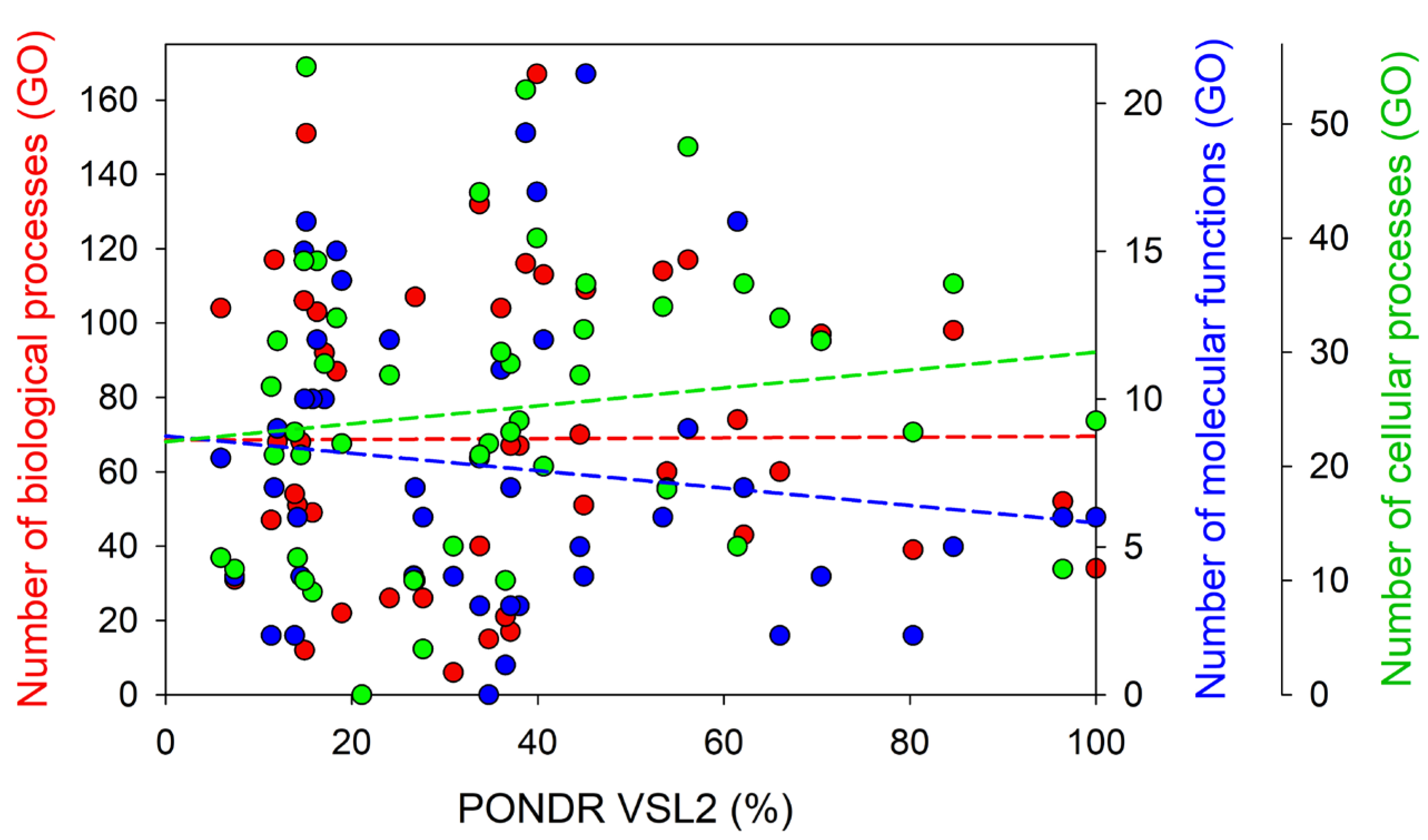
3.1. Global Disorder Analysis of Host Proteins Entrapped in RABV Particles
3.2. Functional Intrinsic Disorder in the Most Disordered Mouse Proteins Found in the Rabies Virus
Neuromodulin (UniProt ID: P06837)
3.3. Global PPI Networks Analysis of the Most Disordered Mouse Proteins Found in the Rabies Virus
3.4. The Roles of Intrinsically Disordered Host Proteins in Viral Immune Evasion and Pathogenesis Enhancement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Functional Intrinsic Disorder in the Most Disordered Mouse Proteins, Chmp4b, DnaJB6, Vps37B, and Wasl, Found in the Rabies Virus
Appendix A.1.1. Charged Multivesicular Body Protein 4b (Chmp4b, UniProt ID: Q9D8B3)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region Type | Region Range | ELM ID | Position |
|---|---|---|---|
| MoRF | 1–12 | LIG_BIR_II_1 | 1–5 |
| LIG_LIR_Nem_3 | 2–7 | ||
| LIG_Pex14_2 | 4–8 | ||
| Droplet-promoting region | 1–22 | LIG_BIR_II_1 | 1–5 |
| LIG_LIR_Nem_3 | 2–7 | ||
| LIG_Pex14_2 | 4–8 | ||
| DOC_WW_Pin1_4 | 18–23 | ||
| MOD_ProDKin_1 | 18–24 | ||
| LIG_FHA_2 | 19–25 | ||
| Region with multiplicity of binding modes | 27–32 | MOD_PKA_2 | 29–35 |
| Region with multiplicity of binding modes | 39–82 | MOD_SUMO_rev_2 | 41–47 |
| TRG_NLS_Bipartite_1 | 55–75 | ||
| DOC_USP7_UBL2_3 | 56–60 | ||
| CLV_PCSK_PC1ET2_1 | 62–64 | ||
| TRG_NLS_MonoExtN_4 | 70–75 | ||
| TRG_NLS_MonoCore_2 | 69–74 | ||
| 70–75 | |||
| TRG_NLS_MonoExtC_3 | 69–74 | ||
| 70–75 | |||
| Aggregation hotspot | 54–62 | DOC_USP7_UBL2_3 | 56–60 |
| CLV_PCSK_PC1ET2_1 | 62–64 | ||
| MoRF | 108–118 | LIG_SH2_STAP1 | 111–115 |
| LIG_WD40_WDR5_VDV_2 | 111–115 | ||
| CLV_PCSK_SKI1_1 | 114–118 | ||
| MoRF | 141–200 | MOD_GSK3_1 | 143–150 |
| 181–188 | |||
| DOC_PP1_RVXF_1 | 149–156 | ||
| LIG_Pex14_2 | 155–159 | ||
| LIG_WD40_WDR5_VDV_2 | 161–168 | ||
| 162–168 | |||
| 163–168 | |||
| 177–183 | |||
| CLV_PCSK_SKI1_1 | 178–182 | ||
| TRG_Pf-PMV_PEXEL_1 | 178–182 | ||
| LIG_SUMO_SIM_par_1 | 179–184 | ||
| MOD_CK2_1 | 181–187 | ||
| MOD_GlcNHglycan | 182–186 | ||
| 183–186 | |||
| LIG_FHA_1 | 186–192 | ||
| LIG_SH3_3 | 186–192 | ||
| 189–195 | |||
| 194–200 | |||
| 197–203 | |||
| DOC_USP7_MATH_1 | 198–202 | ||
| Region with multiplicity of binding modes | 183–190 | LIG_WD40_WDR5_VDV_2 | 177–183 |
| TRG_Pf-PMV_PEXEL_1 | 178–182 | ||
| LIG_SUMO_SIM_par_1 | 179–184 | ||
| MOD_CK2_1 | 181–187 | ||
| MOD_GlcNHglycan | 182–186 | ||
| 183–186 | |||
| LIG_FHA_1 | 186–192 | ||
| LIG_SH3_3 | 186–192 | ||
| 189–195 | |||
| Region with multiplicity of binding modes | 197–207 | DOC_USP7_MATH_1 | 198–202 |
| LIG_SH3_2 | 200–205 | ||
| CLV_PCSK_SKI1_1 | 202–206 | ||
| DOC_USP7_UBL2_3 | 202–206 | ||
| Aggregation hotspot | 197–207 | DOC_USP7_MATH_1 | 198–202 |
| LIG_SH3_2 | 200–205 | ||
| CLV_PCSK_SKI1_1 | 202–206 | ||
| DOC_USP7_UBL2_3 | 202–206 | ||
| Droplet-promoting region | 190–224 | LIG_FHA_1 | 186–192 |
| LIG_SH3_3 | 186–192 | ||
| 189–195 | |||
| 194–200 | |||
| 197–203 | |||
| DOC_USP7_MATH_1 | 198–202 | ||
| LIG_SH3_2 | 200–205 | ||
| CLV_PCSK_SKI1_1 | 202–206 | ||
| DOC_USP7_UBL2_3 | 202–206 | ||
| LIG_SH3_4 | 202–209 | ||
| TRG_NESrev_CRM1_2 | 208–217 | ||
| 209–217 | |||
| 210–217 | |||
| 211–217 | |||
| 212–217 | |||
| Aggregation hotspot | 211–217 | TRG_NESrev_CRM1_2 | 208–217 |
| 209–217 | |||
| 210–217 | |||
| 211–217 | |||
| 212–217 | |||
| MoRF | 214–224 | TRG_NESrev_CRM1_2 | 208–217 |
| 209–217 | |||
| 210–217 | |||
| 211–217 | |||
| 212–217 | |||
| MOD_SUMO_rev_2 | 212–217 |
Appendix A.1.2. DnaJ Homolog Subfamily B Member 6 (DNAJB6; UniProt ID: O54946)

| Region Type | Region Range | ELM ID | Position |
|---|---|---|---|
| Region with multiplicity of binding modes | 14–23 | DOC_WW_Pin1_4 | 12–17 |
| CLV_NRD_NRD_1 | 23–25 | ||
| Region with multiplicity of binding modes | 39–55 | CLV_NRD_NRD_1 | 43–45 |
| CLV_PCSK_SKI1_1 | 44–48 | ||
| Droplet-promoting region | 58–94 | LIG_LIR_Nem_3 | 63–68 |
| DOC_WW_Pin1_4 | 83–88 | ||
| DOC_PP4_FxxP_1 | 84–87 | ||
| Region with multiplicity of binding modes | 57–69 | TRG_Pf-PMV_PEXEL_1 | 62–66 |
| LIG_LIR_Nem_3 | 63–68 | ||
| Aggregation hotspot | 58–69 | TRG_Pf-PMV_PEXEL_1 | 62–66 |
| LIG_LIR_Nem_3 | 63–68 | ||
| Droplet-promoting region | 58–94 | TRG_Pf-PMV_PEXEL_1 | 62–66 |
| DOC_PP4_FxxP_1 | 84–87 | ||
| 94–97 | |||
| Region with multiplicity of binding modes | 83–90 | DOC_WW_Pin1_4 | 83–88 |
| MOD_ProDKin_1 | 83–89 | ||
| DOC_PP4_FxxP_1 | 84–87 | ||
| Aggregation hotspot | 83–90 | DOC_WW_Pin1_4 | 83–88 |
| MOD_ProDKin_1 | 83–89 | ||
| DOC_PP4_FxxP_1 | 84–87 | ||
| Region with multiplicity of binding modes | 93–131 | DOC_PP4_FxxP_1 | 84–87 |
| 94–97 | |||
| CLV_PCSK_SKI1_1 | 102–106 | ||
| Aggregation hotspot | 105–114 | LIG_BRCT_BRCA1_1 | 111–115 |
| LIG_AP2alpha_2 | 109–111 | ||
| Aggregation hotspot | 119–131 | DOC_PP4_FxxP_1 | 116–119 |
| LIG_AP2alpha_1 | 116–120 | ||
| 120–124 | |||
| LIG_AP2alpha_2 | 118–120 | ||
| CLV_NRD_NRD_1 | 127–129 | ||
| CLV_PCSK_KEX2_1 | 127–129 | ||
| Droplet-promoting region | 119–185 | DOC_PP4_FxxP_1 | 116–119 |
| LIG_AP2alpha_1 | 116–120 | ||
| 120–124 | |||
| CLV_NRD_NRD_1 | 127–129 | ||
| CLV_PCSK_KEX2_1 | 127–129 | ||
| LIG_Arc_Nlobe_1 | 148–152 | ||
| 155–120 | |||
| OC_USP7_MATH_1 | 164–168 | ||
| LIG_BRCT_BRCA1_1 | 177–181 | ||
| Aggregation hotspot | 156–185 | LIG_Arc_Nlobe_1 | 155–159 |
| DOC_WW_Pin1_4 | 160–165 | ||
| OC_USP7_MATH_1 | 164–168 | ||
| LIG_BRCT_BRCA1_1 | 177–181 | ||
| Region with multiplicity of binding modes | 156–203 | OC_USP7_MATH_1 | 164–168 |
| LIG_BRCT_BRCA1_1 | 177–181 | ||
| CLV_PCSK_SKI1_1 | 202–206 | ||
| DOC_USP7_UBL2_3 | 203–207 | ||
| Region with multiplicity of binding modes | 206–211 | CLV_PCSK_SKI1_1 | 202–206 |
| DOC_USP7_UBL2_3 | 203–207 | ||
| CLV_PCSK_KEX2_1 | 207–209 | ||
| MoRF | 223–278 | CLV_NRD_NRD_1 | 245–247 |
| CLV_PCSK_SKI1_1 | 226–230 | ||
| Region with multiplicity of binding modes | 227–237 | CLV_PCSK_SKI1_1 | 226–230 |
| Droplet-promoting region | 233–365 | CLV_NRD_NRD_1 | 245–247 |
| DEG_ODPH_VHL_1 | 253–264 | ||
| DOC_USP7_MATH_1 | 291–295 | ||
| 293–297 | |||
| 334–338 | |||
| DEG_SCF_FBW7_1 | 271–278 | ||
| 273–278 | |||
| 275–282 | |||
| 277–282 | |||
| 287–294 | |||
| DOC_USP7_UBL2_3 | 310–314 | ||
| 341–345 | |||
| 348–352 | |||
| 352–356 | |||
| 358–362 | |||
| Region with multiplicity of binding modes | 241–250 | CLV_NRD_NRD_1 | 245–247 |
| DOC_CKS1_1 | 248–253 | ||
| Aggregation hotspot | 241–250 | CLV_NRD_NRD_1 | 245–247 |
| DOC_CKS1_1 | 248–253 | ||
| MoRF | 282–298 | DEG_SCF_FBW7_1 | 275–282 |
| 277–282 | |||
| 287–294 | |||
| DOC_WW_Pin1_4 | 279–284 | ||
| 287–292 | |||
| DOC_USP7_MATH_1 | 291–295 | ||
| 293–297 | |||
| LIG_WD40_WDR5_VDV_2 | 290–295 | ||
| Region with multiplicity of binding modes | 316–323 | MOD_CK2_1 | 312–318 |
| DOC_ANK_TNKS_1 | 323–330 | ||
| Aggregation hotspot | 316–323 | MOD_CK2_1 | 312–318 |
| DOC_ANK_TNKS_1 | 323–330 | ||
| Aggregation hotspot | 345–353 | OC_USP7_UBL2_3 | 341–345 |
| 348–352 | |||
| 352–356 | |||
| DOC_USP7_UBL2_3 | 341–345 | ||
| 348–352 | |||
| 352–356 | |||
| TRG_NLS_Bipartite_1 | 345–361 | ||
| 346–261 | |||
| 347–361 | |||
| CLV_NRD_NRD_1 | 345–347 | ||
| CLV_PCSK_KEX2_1 | 345–347 | ||
| Region with multiplicity of binding modes | 345–353 | OC_USP7_UBL2_3 | 341–345 |
| 348–352 | |||
| 352–356 | |||
| DOC_USP7_UBL2_3 | 341–345 | ||
| 348–352 | |||
| 352–356 | |||
| TRG_NLS_Bipartite_1 | 345–361 | ||
| 346–261 | |||
| 347–361 | |||
| CLV_NRD_NRD_1 | 345–347 | ||
| CLV_PCSK_KEX2_1 | 345–347 | ||
| MoRF | 305–365 | DOC_USP7_UBL2_3 | 310–314 |
| 341–345 | |||
| 348–352 | |||
| 352–356 | |||
| 358–362 | |||
| OC_USP7_UBL2_3 | 341–345 | ||
| 348–352 | |||
| 352–356 | |||
| 368–362 | |||
| TRG_NLS_Bipartite_1 | 345–361 | ||
| 346–261 | |||
| 347–361 | |||
| DOC_USP7_MATH_1 | 334–338 | ||
| CLV_NRD_NRD_1 | 345–347 | ||
| CLV_PCSK_KEX2_1 | 345–347 |
Appendix A.1.3. Vacuolar Protein Sorting-Associated Protein 37B (Vps37B, UniProt ID: Q8R0J7)


Appendix A.1.4. Actin Nucleation-Promoting Factor Wasl (UniProt ID: Q91YD9)


References
- Hemachudha, T.; Laothamatas, J.; Rupprecht, C.E. Human rabies: A disease of complex neuropathogenetic mechanisms and diagnostic challenges. Lancet Neurol. 2002, 1, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, J.E. Rabies. Available online: https://www.merckmanuals.com/professional/neurologic-disorders/brain-infections/rabies (accessed on 18 March 2024).
- Pieracci, E.G.; Pearson, C.M.; Wallace, R.M.; Blanton, J.D.; Whitehouse, E.R.; Ma, X.; Stauffer, K.; Chipman, R.B.; Olson, V. Vital Signs: Trends in Human Rabies Deaths and Exposures—United States, 1938–2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Estimating the global burden of endemic canine rabies. PLoS Negl. Trop. Dis. 2015, 9, e0003709. [Google Scholar] [CrossRef] [PubMed]
- Brunker, K.; Mollentze, N. Rabies Virus. Trends Microbiol. 2018, 26, 886–887. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, C.E. Rhabdoviruses: Rabies Virus. In Medical Microbiology, 4th ed.; Baron, S., Ed.; The University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Horwitz, J.A.; Jenni, S.; Harrison, S.C.; Whelan, S.P.J. Structure of a rabies virus polymerase complex from electron cryo-microscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Riedel, C.; Vasishtan, D.; Prazak, V.; Ghanem, A.; Conzelmann, K.K.; Rumenapf, T. Cryo EM structure of the rabies virus ribonucleoprotein complex. Sci. Rep. 2019, 9, 9639. [Google Scholar] [CrossRef] [PubMed]
- Warrell, M.J.; Warrell, D.A. Rabies: The clinical features, management and prevention of the classic zoonosis. Clin. Med. 2015, 15, 78. [Google Scholar] [CrossRef]
- Singh, R.; Singh, K.P.; Cherian, S.; Saminathan, M.; Kapoor, S.; Manjunatha Reddy, G.; Panda, S.; Dhama, K. Rabies–epidemiology, pathogenesis, public health concerns and advances in diagnosis and control: A comprehensive review. Vet. Q. 2017, 37, 212–251. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.M.; Rall, G.F.; Schnell, M.J. Everything You Always Wanted to Know About Rabies Virus (But Were Afraid to Ask). Annu. Rev. Virol. 2015, 2, 451–471. [Google Scholar] [CrossRef]
- Potratz, M.; Zaeck, L.M.; Weigel, C.; Klein, A.; Freuling, C.M.; Muller, T.; Finke, S. Neuroglia infection by rabies virus after anterograde virus spread in peripheral neurons. Acta Neuropathol. Commun. 2020, 8, 199. [Google Scholar] [CrossRef]
- Piccinotti, S.; Whelan, S.P. Rabies Internalizes into Primary Peripheral Neurons via Clathrin Coated Pits and Requires Fusion at the Cell Body. PLoS Pathog. 2016, 12, e1005753. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, Y.; Tuffereau, C.; Segretain, D.; Knossow, M.; Flamand, A. Reversible conformational changes and fusion activity of rabies virus glycoprotein. J. Virol. 1991, 65, 4853–4859. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, X.; Vidy, A.; Pomier, C.; Obiang, L.; Harper, F.; Gaudin, Y.; Blondel, D. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: Evidence that NBs are sites of viral transcription and replication. J. Virol. 2009, 83, 7948–7958. [Google Scholar] [CrossRef] [PubMed]
- Negri, A. Contributo allo Studio Dell’eziologia della Rabbia; Tipografia e Legatoria Cooperativa: Pavia, Italy, 1905. [Google Scholar]
- Nevers, Q.; Albertini, A.A.; Lagaudriere-Gesbert, C.; Gaudin, Y. Negri bodies and other virus membrane-less replication compartments. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118831. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.; Camporeale, G.; Salgueiro, M.; Borkosky, S.S.; Visentin, A.; Peralta-Martinez, R.; Loureiro, M.E.; de Prat-Gay, G. Deconstructing virus condensation. PLoS Pathog. 2021, 17, e1009926. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Green, T.J. Transcriptional Control and mRNA Capping by the GDP Polyribonucleotidyltransferase Domain of the Rabies Virus Large Protein. Viruses 2019, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Wunner, W.H.; Conzelmann, K.K. Rabies virus. In Rabies, 3rd ed.; Jackson, A.C., Ed.; Academic Press/Elsevier: Oxford, UK, 2013; pp. 17–60. [Google Scholar]
- Burnie, J.; Guzzo, C. The Incorporation of Host Proteins into the External HIV-1 Envelope. Viruses 2019, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci. 2013, 22, 693–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Feng, Y.; Tu, Z.; Lou, Z.; Tu, C. Proteomic Profiling of Purified Rabies Virus Particles. Virol. Sin. 2020, 35, 143–155. [Google Scholar] [CrossRef]
- Lahaye, X.; Vidy, A.; Fouquet, B.; Blondel, D. Hsp70 protein positively regulates rabies virus infection. J. Virol. 2012, 86, 4743–4751. [Google Scholar] [CrossRef]
- Chen, B.J.; Lamb, R.A. Mechanisms for enveloped virus budding: Can some viruses do without an ESCRT? Virology 2008, 372, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Bhattacharya, S.; Achuthan, S.; Behal, A.; Jolly, M.K.; Kotnala, S.; Mohanty, A.; Rangarajan, G.; Salgia, R.; Uversky, V. Intrinsically Disordered Proteins: Critical Components of the Wetware. Chem. Rev. 2022, 122, 6614–6633. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, T.; Romero, P.R.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in the Protein Data Bank. J. Biomol. Struct. Dyn. 2007, 24, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J. Making Sense of Intrinsically Disordered Proteins. Biophys. J. 2016, 110, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef]
- Dayhoff, G.W., 2nd; Uversky, V.N. Rapid prediction and analysis of protein intrinsic disorder. Protein Sci. 2022, 31, e4496. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Tosatto, S.C. Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Conte, A.D.; Mehdiabadi, M.; Bouhraoua, A.; Miguel Monzon, A.; Tosatto, S.C.E.; Piovesan, D. Critical assessment of protein intrinsic disorder prediction (CAID)—Results of round 2. Proteins 2023, 91, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Puntervoll, P.; Linding, R.; Gemund, C.; Chabanis-Davidson, S.; Mattingsdal, M.; Cameron, S.; Martin, D.M.; Ausiello, G.; Brannetti, B.; Costantini, A.; et al. ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res. 2003, 31, 3625–3630. [Google Scholar] [CrossRef]
- Gould, C.M.; Diella, F.; Via, A.; Puntervoll, P.; Gemund, C.; Chabanis-Davidson, S.; Michael, S.; Sayadi, A.; Bryne, J.C.; Chica, C.; et al. ELM: The status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 2010, 38, D167–D180. [Google Scholar] [CrossRef] [PubMed]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, H.; Michael, S.; Weatheritt, R.J.; Davey, N.E.; Van Roey, K.; Altenberg, B.; Toedt, G.; Uyar, B.; Seiler, M.; Budd, A.; et al. ELM—The database of eukaryotic linear motifs. Nucleic Acids Res. 2012, 40, D242–D251. [Google Scholar] [CrossRef] [PubMed]
- Gouw, M.; Samano-Sanchez, H.; Van Roey, K.; Diella, F.; Gibson, T.J.; Dinkel, H. Exploring Short Linear Motifs Using the ELM Database and Tools. Curr. Protoc. Bioinform. 2017, 58, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gouw, M.; Michael, S.; Samano-Sanchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Calyseva, J.; et al. ELM-the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2020, 48, D296–D306. [Google Scholar] [CrossRef]
- Kumar, M.; Michael, S.; Alvarado-Valverde, J.; Zeke, A.; Lazar, T.; Glavina, J.; Nagy-Kanta, E.; Donagh, J.M.; Kalman, Z.E.; Pascarelli, S.; et al. ELM-the Eukaryotic Linear Motif resource-2024 update. Nucleic Acids Res. 2024, 52, D442–D455. [Google Scholar] [CrossRef]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc. Natl. Acad. Sci. USA 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Hatos, A.; Tosatto, S.C.E.; Vendruscolo, M.; Fuxreiter, M. FuzDrop on AlphaFold: Visualizing the sequence-dependent propensity of liquid-liquid phase separation and aggregation of proteins. Nucleic Acids Res. 2022, 50, W337–W344. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. Biosyst. 2008, 4, 328–340. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J. Cell Biochem. 2011, 112, 3256–3267. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Pac. Symp. Biocomput. 2012, 128–139. [Google Scholar]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Multitude of binding modes attainable by intrinsically disordered proteins: A portrait gallery of disorder-based complexes. Chem. Soc. Rev. 2011, 40, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef] [PubMed]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem.-Int. Ed. 1998, 37, 2755–2794. [Google Scholar] [CrossRef]
- Schulz, G.E. Nucleotide Binding Proteins. In Molecular Mechanism of Biological Recognition; Balaban, M., Ed.; Elsevier/North-Holland Biomedical Press: New York, NY, USA, 1979; pp. 79–94. [Google Scholar]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 2002, 62, 25–49. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Meador, W.E.; Means, A.R.; Quiocho, F.A. Modulation of calmodulin plasticity in molecular recognition on the basis of X-ray structures. Science 1993, 262, 1718–1721. [Google Scholar] [CrossRef] [PubMed]
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 11504–11509. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: Which way to go? Cell Mol. Life Sci. 2003, 60, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets: The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Dajani, R.; Fraser, E.; Roe, S.M.; Yeo, M.; Good, V.M.; Thompson, V.; Dale, T.C.; Pearl, L.H. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. Embo J. 2003, 22, 494–501. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.L.; Oldfield, C.J.; Xue, B.; Meng, J.; Huang, F.; Romero, P.; Uversky, V.N.; Dunker, A.K. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2013, 22, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008, 9 (Suppl. S1), S1. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hazy, E.; Tompa, P. Limitations of induced folding in molecular recognition by intrinsically disordered proteins. Chemphyschem 2009, 10, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.; Aivazian, D.; Stern, L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry 2004, 43, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.B.; Zhuravleva, A.V.; Orekhov, V.Y. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie 2007, 89, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Permyakov, S.E.; Millett, I.S.; Doniach, S.; Permyakov, E.A.; Uversky, V.N. Natively unfolded C-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins 2003, 53, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M. Fuzziness: Linking regulation to protein dynamics. Mol. Biosyst. 2012, 8, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P. Fuzzy complexes: A more stochastic view of protein function. Adv. Exp. Med. Biol. 2012, 725, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015, 589, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006, 580, 2041–2045. [Google Scholar] [CrossRef]
- Ekman, D.; Light, S.; Bjorklund, A.K.; Elofsson, A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol. 2006, 7, R45. [Google Scholar] [CrossRef]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006, 2, e100. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res. 2006, 5, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.P.; Dash, D. Intrinsic disorder in yeast transcriptional regulatory network. Proteins 2007, 68, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.P.; Ganapathi, M.; Dash, D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins 2007, 66, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Turoverov, K.K.; Kuznetsova, I.M.; Fonin, A.V.; Darling, A.L.; Zaslavsky, B.Y.; Uversky, V.N. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem. Sci. 2019, 44, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef]
- Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. 2015, 589, 15–22. [Google Scholar] [CrossRef]
- Brangwynne, C.P. Phase transitions and size scaling of membrane-less organelles. J. Cell Biol. 2013, 203, 875–881. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2016, 44, 18–30. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Zuber, M.X.; Strittmatter, S.M.; Fishman, M.C. A membrane-targeting signal in the amino terminus of the neuronal protein GAP-43. Nature 1989, 341, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Chapman, E.R.; Storm, D.R. Targeting of neuromodulin (GAP-43) fusion proteins to growth cones in cultured rat embryonic neurons. Neuron 1991, 6, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fisher, D.A.; Storm, D.R. Intracellular sorting of neuromodulin (GAP-43) mutants modified in the membrane targeting domain. J. Neurosci. 1994, 14, 5807–5817. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chichili, V.P.; Zhong, L.; Tang, X.; Velazquez-Campoy, A.; Sheu, F.S.; Seetharaman, J.; Gerges, N.Z.; Sivaraman, J. Structural basis for the interaction of unstructured neuron specific substrates neuromodulin and neurogranin with Calmodulin. Sci. Rep. 2013, 3, 1392. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Mani, S.; Donovan, S.L.; Schwob, J.E.; Meiri, K.F. Growth-associated protein-43 is required for commissural axon guidance in the developing vertebrate nervous system. J. Neurosci. 2002, 22, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.R.; Au, D.; Alexander, K.A.; Nicolson, T.A.; Storm, D.R. Characterization of the calmodulin binding domain of neuromodulin. Functional significance of serine 41 and phenylalanine 42. J. Biol. Chem. 1991, 266, 207–213. [Google Scholar] [CrossRef]
- Katuwawala, A.; Peng, Z.; Yang, J.; Kurgan, L. Computational Prediction of MoRFs, Short Disorder-to-order Transitioning Protein Binding Regions. Comput. Struct. Biotechnol. J. 2019, 17, 454–462. [Google Scholar] [CrossRef]
- Marnik, E.A.; Updike, D.L. Membraneless organelles: P granules in Caenorhabditis elegans. Traffic 2019, 20, 373–379. [Google Scholar] [CrossRef]
- Damian, R.T. Molecular mimicry: Antigen sharing by parasite and host and its consequences. Am. Nat. 1964, 98, 129–149. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Human Immunodeficiency Virus Proteins Mimic Human T Cell Receptors Inducing Cross-Reactive Antibodies. Int. J. Mol. Sci. 2017, 18, 2091. [Google Scholar] [CrossRef]
- Sorci, G.; Cornet, S.; Faivre, B. Immune evasion, immunopathology and the regulation of the immune system. Pathogens 2013, 2, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Restrepo-Jimenez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramirez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S. Virus-Receptor Interactions: The Key to Cellular Invasion. J. Mol. Biol. 2018, 430, 2590–2611. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.A.; Willberg, C.B.; Paul, K. Immune evasion by viruses. In eLS 2013; John Wiley & Sons, Ltd.: Chichester, UK, 2013. [Google Scholar] [CrossRef]
- Bule, M.; Khan, F.; Niaz, K. Antivirals: Past, present and future. In Recent Advvances in Animal Virology; Malik, Y., Singh, R., Yadav, M., Eds.; Springer: Singapore, 2019; pp. 425–446. [Google Scholar] [CrossRef]
- Morris, O.M.; Torpey, J.H.; Isaacson, R.L. Intrinsically disordered proteins: Modes of binding with emphasis on disordered domains. Open Biol. 2021, 11, 210222. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bennett, T.M.; Shiels, A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation 2019, 109, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Teis, D. The ESCRT machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Freed, E.O.; van Engelenburg, S.B. A Consensus View of ESCRT-Mediated Human Immunodeficiency Virus Type 1 Abscission. Annu. Rev. Virol. 2017, 4, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sharma, A.; Patil, A.; Tsunoda, T. Discovering MoRFs by trisecting intrinsically disordered protein sequence into terminals and middle regions. BMC Bioinform. 2019, 19, 378. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.; Dent, K.C.; Zeev-Ben-Mordehai, T.; Grange, M.; Bosse, J.B.; Whittle, C.; Klupp, B.G.; Siebert, C.A.; Vasishtan, D.; Bauerlein, F.J.; et al. Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell 2015, 163, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, Z.; Cao, Y.; Zhang, S.; Li, H.; Huang, Y.; Ding, Y.Q.; Liu, X. The Hsp40 family chaperone protein DnaJB6 enhances Schlafen1 nuclear localization which is critical for promotion of cell-cycle arrest in T-cells. Biochem. J. 2008, 413, 239–250. [Google Scholar] [CrossRef]
- Sarparanta, J.; Jonson, P.H.; Golzio, C.; Sandell, S.; Luque, H.; Screen, M.; McDonald, K.; Stajich, J.M.; Mahjneh, I.; Vihola, A.; et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 2012, 44, 450–455. [Google Scholar] [CrossRef]
- Hageman, J.; Rujano, M.A.; van Waarde, M.A.; Kakkar, V.; Dirks, R.P.; Govorukhina, N.; Oosterveld-Hut, H.M.; Lubsen, N.H.; Kampinga, H.H. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 2010, 37, 355–369. [Google Scholar] [CrossRef]
- Osterlund, N.; Frankel, R.; Carlsson, A.; Thacker, D.; Karlsson, M.; Matus, V.; Graslund, A.; Emanuelsson, C.; Linse, S. The C-terminal domain of the antiamyloid chaperone DNAJB6 binds to amyloid-beta peptide fibrils and inhibits secondary nucleation. J. Biol. Chem. 2023, 299, 105317. [Google Scholar] [CrossRef]
- Kuiper, E.F.E.; Gallardo, P.; Bergsma, T.; Mari, M.; Kolbe Musskopf, M.; Kuipers, J.; Giepmans, B.N.G.; Steen, A.; Kampinga, H.H.; Veenhoff, L.M.; et al. The chaperone DNAJB6 surveils FG-nucleoporins and is required for interphase nuclear pore complex biogenesis. Nat. Cell Biol. 2022, 24, 1584–1594. [Google Scholar] [CrossRef]
- Izawa, I.; Nishizawa, M.; Ohtakara, K.; Ohtsuka, K.; Inada, H.; Inagaki, M. Identification of Mrj, a DnaJ/Hsp40 family protein, as a keratin 8/18 filament regulatory protein. J. Biol. Chem. 2000, 275, 34521–34527. [Google Scholar] [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Szymanska, E.; Budick-Harmelin, N.; Miaczynska, M. Endosomal “sort” of signaling control: The role of ESCRT machinery in regulation of receptor-mediated signaling pathways. Semin. Cell Dev. Biol. 2018, 74, 11–20. [Google Scholar] [CrossRef]
- Olmos, Y.; Carlton, J.G. The ESCRT machinery: New roles at new holes. Curr. Opin. Cell Biol. 2016, 38, 1–11. [Google Scholar] [CrossRef]
- Hurley, J.H. ESCRTs are everywhere. EMBO J. 2015, 34, 2398–2407. [Google Scholar] [CrossRef]
- Wunderley, L.; Brownhill, K.; Stefani, F.; Tabernero, L.; Woodman, P. The molecular basis for selective assembly of the UBAP1-containing endosome-specific ESCRT-I complex. J. Cell Sci. 2014, 127, 663–672. [Google Scholar] [CrossRef]
- Stefani, F.; Zhang, L.; Taylor, S.; Donovan, J.; Rollinson, S.; Doyotte, A.; Brownhill, K.; Bennion, J.; Pickering-Brown, S.; Woodman, P. UBAP1 is a component of an endosome-specific ESCRT-I complex that is essential for MVB sorting. Curr. Biol. 2011, 21, 1245–1250. [Google Scholar] [CrossRef]
- Kolmus, K.; Erdenebat, P.; Szymanska, E.; Stewig, B.; Goryca, K.; Derezinska-Wolek, E.; Szumera-Cieckiewicz, A.; Brewinska-Olchowik, M.; Piwocka, K.; Prochorec-Sobieszek, M.; et al. Concurrent depletion of Vps37 proteins evokes ESCRT-I destabilization and profound cellular stress responses. J. Cell Sci. 2021, 134, 1245–1250. [Google Scholar] [CrossRef]
- Bajorek, M.; Morita, E.; Skalicky, J.J.; Morham, S.G.; Babst, M.; Sundquist, W.I. Biochemical analyses of human IST1 and its function in cytokinesis. Mol. Biol. Cell 2009, 20, 1360–1373. [Google Scholar] [CrossRef]
- Derry, J.M.; Ochs, H.D.; Francke, U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell 1994, 78, 635–644. [Google Scholar] [CrossRef]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef]
- Alekhina, O.; Burstein, E.; Billadeau, D.D. Cellular functions of WASP family proteins at a glance. J. Cell Sci. 2017, 130, 2235–2241. [Google Scholar] [CrossRef]
- Goley, E.D.; Welch, M.D. The ARP2/3 complex: An actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006, 7, 713–726. [Google Scholar] [CrossRef]
- Burianek, L.E.; Soderling, S.H. Under lock and key: Spatiotemporal regulation of WASP family proteins coordinates separate dynamic cellular processes. Semin. Cell Dev. Biol. 2013, 24, 258–266. [Google Scholar] [CrossRef]
- Stanganello, E.; Hagemann, A.I.; Mattes, B.; Sinner, C.; Meyen, D.; Weber, S.; Schug, A.; Raz, E.; Scholpp, S. Filopodia-based Wnt transport during vertebrate tissue patterning. Nat. Commun. 2015, 6, 5846. [Google Scholar] [CrossRef]
- Verboon, J.M.; Sugumar, B.; Parkhurst, S.M. Wiskott-Aldrich syndrome proteins in the nucleus: aWASH with possibilities. Nucleus 2015, 6, 349–359. [Google Scholar] [CrossRef]







| ID | Description | p-Value |
|---|---|---|
| Biological Process (Gene Ontology) | ||
| GO:0051128 | Regulation of cellular component organization | 4.73 × 10−14 |
| GO:0051049 | Regulation of transport | 1.28 × 10−12 |
| GO:0032879 | Regulation of localization | 4.85 × 10−12 |
| GO:0051050 | Positive regulation of transport | 8.84 × 10−12 |
| GO:0008104 | Protein localization | 8.84 × 10−12 |
| Molecular Function (Gene Ontology) | ||
| GO:0044877 | Protein-containing complex binding | 1.53 × 10−07 |
| GO:0005515 | Protein binding | 3.76 × 10−07 |
| GO:0003925 | G protein activity | 6.11 × 10−05 |
| GO:0005488 | Binding | 6.56 × 10−05 |
| GO:0019904 | Protein domain-specific binding | 0.00014 |
| Cellular Component (Gene Ontology) | ||
| GO:0031982 | Vesicle | 8.04 × 10−13 |
| GO:0042470 | Melanosome | 1.16 × 10−12 |
| GO:0031410 | Cytoplasmic vesicle | 5.80 × 10−12 |
| GO:0005829 | Cytosol | 1.83 × 10−11 |
| GO:0005768 | Endosome | 2.11 × 10−11 |
| Protein | ID | Description | p-Value |
|---|---|---|---|
| Neuromodulin | Biological Process (Gene Ontology) | ||
| GO:0022008 | Neurogenesis | 2.34 × 10−52 | |
| GO:0007399 | Nervous system development | 2.34 × 10−52 | |
| GO:0048699 | Generation of neurons | 1.74 × 10−48 | |
| GO:0030182 | Neuron differentiation | 7.50 × 10−47 | |
| GO:0048666 | Neuron development | 2.92 × 10−43 | |
| Molecular Function (Gene Ontology) | |||
| GO:0005515 | Protein binding | 1.35 × 10−25 | |
| GO:0005488 | Binding | 6.32 × 10−18 | |
| GO:0042802 | Identical protein binding | 1.38 × 10−10 | |
| GO:0005102 | Signaling receptor binding | 5.78 × 10−10 | |
| GO:0044877 | Protein-containing complex binding | 9.88 × 10−10 | |
| Cellular Component (Gene Ontology) | |||
| GO:0030424 | Axon | 2.24 × 10−59 | |
| GO:0036477 | Somatodendritic compartment | 3.44 × 10−50 | |
| GO:0043005 | Neuron projection | 3.44 × 10−50 | |
| GO:0044297 | Cell body | 2.06 × 10−48 | |
| GO:0043025 | Neuronal cell body | 2.87 × 10−46 | |
| Chmp4b | Biological Process (Gene Ontology) | ||
| GO:0043162 | Ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway | 6.73 × 10−43 | |
| GO:0071985 | Multivesicular body sorting pathway | 1.62 × 10−33 | |
| GO:0007034 | Vacuolar transport | 2.43 × 10−33 | |
| GO:0032509 | Endosome transport via multivesicular body sorting pathway | 8.11 × 10−33 | |
| GO:0045324 | Late endosome to vacuole transport | 5.55 × 10−32 | |
| Molecular Function (Gene Ontology) | |||
| GO:0005212 | Structural constituent of eye lens | 4.36 × 10−20 | |
| GO:0005198 | Structural molecule activity | 1.15 × 10−18 | |
| GO:0005200 | Structural constituent of cytoskeleton | 8.50 × 10−15 | |
| GO:0005525 | GTP binding | 3.97 × 10−09 | |
| GO:0044389 | Ubiquitin-like protein ligase binding | 4.28 × 10−07 | |
| Cellular Component (Gene Ontology) | |||
| GO:0036452 | ESCRT complex | 3.59 × 10−45 | |
| GO:0005768 | Endosome | 7.27 × 10−29 | |
| GO:0010008 | Endosome membrane | 3.37 × 10−28 | |
| GO:0005770 | Late endosome | 5.21 × 10−27 | |
| GO:0031902 | Late endosome membrane | 1.09 × 10−26 | |
| DnaJB6 | Biological Process (Gene Ontology) | ||
| GO:0006457 | Protein folding | 6.61 × 10−43 | |
| GO:0061077 | Chaperone-mediated protein folding | 8.46 × 10−27 | |
| GO:0042026 | Protein refolding | 1.27 × 10−25 | |
| GO:0035966 | Response to topologically incorrect protein | 1.27 × 10−20 | |
| GO:0006986 | Response to unfolded protein | 2.60 × 10−20 | |
| Molecular Function (Gene Ontology) | |||
| GO:0051082 | Unfolded protein binding | 1.35 × 10−40 | |
| GO:0044183 | Protein folding chaperone | 3.88 × 10−38 | |
| GO:0140662 | ATP-dependent protein folding chaperone | 2.63 × 10−31 | |
| GO:0031072 | Heat shock protein binding | 6.85 × 10−24 | |
| GO:0051087 | Chaperone binding | 1.15 × 10−18 | |
| Cellular Component (Gene Ontology) | |||
| GO:0005737 | Cytoplasm | 8.10 × 10−14 | |
| GO:0101031 | Chaperone complex | 1.59 × 10−13 | |
| GO:0005829 | Cytosol | 1.45 × 10−11 | |
| GO:0005759 | Mitochondrial matrix | 2.49 × 10−10 | |
| GO:0005739 | Mitochondrion | 4.02 × 10−09 | |
| Vps37B | Biological Process (Gene Ontology) | ||
| GO:0043162 | Ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway | 1.09 × 10−52 | |
| GO:0007034 | Vacuolar transport | 2.94 × 10−45 | |
| GO:0071985 | Multivesicular body sorting pathway | 3.42 × 10−41 | |
| GO:0032509 | Endosome transport via multivesicular body sorting pathway | 9.64 × 10−40 | |
| GO:0045324 | Late endosome to vacuole transport | 2.43 × 10−38 | |
| Molecular Function (Gene Ontology) | |||
| GO:0043130 | Ubiquitin binding | 3.22 × 10−11 | |
| GO:0005515 | Protein binding | 2.21 × 10−06 | |
| GO:0031386 | Protein tag | 4.31 × 10−05 | |
| GO:0019904 | Protein domain-specific binding | 4.37 × 10−05 | |
| GO:0090541 | MIT domain binding | 0.00019 | |
| Cellular Component (Gene Ontology) | |||
| GO:0036452 | ESCRT complex | 9.78 × 10−58 | |
| GO:0010008 | Endosome membrane | 8.71 × 10−42 | |
| GO:0031902 | Late endosome membrane | 7.17 × 10−40 | |
| GO:0005770 | Late endosome | 3.41 × 10−39 | |
| GO:0005768 | Endosome | 1.41 × 10−37 | |
| Wasl | Biological Process (Gene Ontology) | ||
| GO:0030029 | Actin filament-based process | 6.73 × 10−119 | |
| GO:0030036 | Actin cytoskeleton organization | 1.35 × 10−117 | |
| GO:0007010 | Cytoskeleton organization | 2.58 × 10−96 | |
| GO:0032956 | Regulation of actin cytoskeleton organization | 1.33 × 10−91 | |
| GO:0032970 | Regulation of actin filament-based process | 2.26 × 10−90 | |
| Molecular Function (Gene Ontology) | |||
| GO:0003779 | Actin binding | 1.01 × 10−71 | |
| GO:0008092 | Cytoskeletal protein binding | 1.68 × 10−71 | |
| GO:0005515 | Protein binding | 7.97 × 10−48 | |
| GO:0051015 | Actin filament binding | 3.00 × 10−40 | |
| GO:0044877 | Protein-containing complex binding | 3.18 × 10−34 | |
| Cellular Component (Gene Ontology) | |||
| GO:0005856 | Cytoskeleton | 4.13 × 10−75 | |
| GO:0031252 | Cell leading edge | 2.82 × 10−74 | |
| GO:0015629 | Actin cytoskeleton | 3.67 × 10−73 | |
| GO:0042995 | Cell projection | 2.16 × 10−63 | |
| GO:0030027 | Lamellipodium | 1.97 × 10−55 | |
| Region Type | Region Range | ELM ID | Position |
|---|---|---|---|
| Droplet-promoting region | 52–277 | LIG_PDZ_Class_3 | 222–227 |
| LIG_WD40_WDR5_VDV_2 | 219–222 | ||
| 218–222 | |||
| 215–222 | |||
| 155–161 | |||
| 154–161 | |||
| 133–137 | |||
| 132–137 | |||
| 131–137 | |||
| 96–99 | |||
| 95–99 | |||
| 64–66 | |||
| 58–64 | |||
| MOD_GlcNHglycan | 209–212 | ||
| 132–135 | |||
| 127–130 | |||
| 85–88 | |||
| 84–88 | |||
| MOD_SUMO_rev_2 | 203–207 | ||
| 200–207 | |||
| 198–207 | |||
| 196–201 | |||
| 193–201 | |||
| 192–201 | |||
| 191–201 | |||
| 154–159 | |||
| 149–159 | |||
| 122–126 | |||
| 118–126 | |||
| CLV_C14_Caspase3-7 | 197–201 | ||
| DOC_USP7_MATH_1 | 207–211 | ||
| 190–194 | |||
| 119–123 | |||
| MOD_CK2_1 | 190–196 | ||
| 142–148 | |||
| MOD_GSK3_1 | 186–193 | ||
| 135–142 | |||
| MOD_PIKK_1 | 190–196 | ||
| LIG_TRAF6_MATH_1 | 184–192 | ||
| DOC_WW_Pin1_4 | 169–174 | ||
| 139–144 | |||
| 93–98 | |||
| MOD_ProDKin_1 | 169–175 | ||
| 139–145 | |||
| 93–99 | |||
| DOC_USP7_UBL2_3 | 153–157 | ||
| MOD_SUMO_for_1 | 152–155 | ||
| 97–100 | |||
| 25–28 | |||
| MOD_CK1_1 | 142–148 | ||
| 128–134 | |||
| 86–92 | |||
| LIG_BIR_III_2 | 118–122 | ||
| MOD_Plk_2-3 | 107–113 | ||
| MOD_CDK_SPK_2 | 93–98 | ||
| MoRF | 102–109 | MOD_Plk_2-3 | 107–113 |
| MoRF | 58–81 | LIG_WD40_WDR5_VDV_2 | 58–64 |
| 63–66 | |||
| Aggregation hotspot | 52–66 | LIG_WD40_WDR5_VDV_2 | 58–64 |
| 63–66 | |||
| MoRF | 1–9 | LIG_UBA3_1 | 1–9 |
| LIG_FHA_1 | 6–12 | ||
| MOD_PKA_2 | 5–11 | ||
| CLV_NRD_NRD_1 | 6–8 | ||
| CLV_PCSK_KEX2_1 | 6–8 | ||
| TRG_ER_diArg_1 | 5–7 | ||
| DEG_Nend_Nbox_1 | 1–3 |
| ID | Description | p-Value |
|---|---|---|
| Biological Process (Gene Ontology) | ||
| GO:0008104 | Protein localization | 0.00017 |
| GO:0051641 | Cellular localization | 0.00017 |
| GO:0019076 | Viral release from host cell | 0.0011 |
| GO:0043162 | Ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway | 0.0013 |
| GO:0016192 | Vesicle-mediated transport | 0.0014 |
| Molecular Function (Gene Ontology) | ||
| GO:0005515 | Protein binding | 0.0470 |
| Cellular Component (Gene Ontology) | ||
| GO:0036452 | ESCRT complex | 0.00071 |
| GO:0031410 | Cytoplasmic vesicle | 0.0011 |
| GO:0005768 | Endosome | 0.0014 |
| GO:0005829 | Cytosol | 0.0048 |
| GO:0000813 | ESCRT I complex | 0.0052 |
| Network | ID | Description | p-Value |
|---|---|---|---|
| Global network | Biological Process (Gene Ontology) | ||
| GO:0016192 | Vesicle-mediated transport | 3.41 × 10−80 | |
| GO:0051641 | Cellular localization | 3.41 × 10−80 | |
| GO:0051128 | Regulation of cellular component organization | 3.22 × 10−76 | |
| GO:0008104 | Protein localization | 2.85 × 10−71 | |
| GO:0051649 | Establishment of localization in cell | 1.38 × 10−65 | |
| Molecular Function (Gene Ontology) | |||
| GO:0005515 | Protein binding | 1.34 × 10−77 | |
| GO:0019904 | Protein domain-specific binding | 8.56 × 10−46 | |
| GO:0008092 | Cytoskeletal protein binding | 1.06 × 10−39 | |
| GO:0005484 | SNAP binding | 2.99 × 10−38 | |
| GO:0005488 | Binding | 5.52 × 10−36 | |
| Cellular Component (Gene Ontology) | |||
| GO:0031982 | Vesicle | 1.74 × 10−73 | |
| GO:0031410 | Cytoplasmic vesicle | 6.16 × 10−66 | |
| GO:0030054 | Cell junction | 6.16 × 10−66 | |
| GO:0042995 | Cell projection | 3.74 × 10−64 | |
| GO:0005488 | Cytoplasm | 3.74 × 10−64 | |
| Cluster 1 | Biological Process (Gene Ontology) | ||
| GO:0005515 | Protein binding | 5.60 × 10−58 | |
| GO:0008092 | Cytoskeletal protein binding | 7.30 × 10−44 | |
| GO:0003779 | Actin binding | 6.12 × 10−43 | |
| GO:0019904 | Protein domain-specific binding | 1.31 × 10−40 | |
| GO:0019899 | Enzyme binding | 1.22 × 10−38 | |
| Molecular Function (Gene Ontology) | |||
| GO:0051128 | Regulation of cellular component organization | 1.80 × 10−65 | |
| GO:0030029 | Actin filament-based process | 1.10 × 10−53 | |
| GO:0044087 | Regulation of cellular component biogenesis | 1.35 × 10−52 | |
| GO:0030036 | Actin cytoskeleton organization | 5.31 × 10−50 | |
| GO:0007010 | Cytoskeleton organization | 5.51 × 10−48 | |
| Cellular Component (Gene Ontology) | |||
| GO:0042995 | Cell projection | 1.61 × 10−62 | |
| GO:0120025 | Plasma membrane bounded cell projection | 2.96 × 10−59 | |
| GO:0005829 | Cytosol | 1.36 × 10−51 | |
| GO:0030054 | Cell junction | 2.26 × 10−50 | |
| GO:0005856 | Cytoskeleton | 6.03 × 10−48 | |
| Cluster 2 | Biological Process (Gene Ontology) | ||
| GO:0043162 | Ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway | 5.74 × 10−45 | |
| GO:0007034 | Vacuolar transport | 4.66 × 10−41 | |
| GO:0071985 | Multivesicular body sorting pathway | 4.84 × 10−37 | |
| GO:0046755 | Viral budding | 2.25 × 10−36 | |
| GO:0032509 | Endosome transport via multivesicular body sorting pathway | 5.26 × 10−36 | |
| Molecular Function (Gene Ontology) | |||
| GO:0043130 | Ubiquitin binding | 6.09 × 10−08 | |
| GO:0005515 | Protein binding | 2.69 × 10−06 | |
| GO:0004459 | L-lactate dehydrogenase activity | 4.33 × 10−06 | |
| GO:0048306 | Calcium-dependent protein binding | 9.28 × 10−06 | |
| GO:0005543 | Phospholipid binding | 5.52 × 10−05 | |
| Cellular Component (Gene Ontology) | |||
| GO:0036452 | ESCRT complex | 1.23 × 10−49 | |
| GO:0010008 | Endosome membrane | 6.18 × 10−43 | |
| GO:0031902 | Late endosome membrane | 1.39 × 10−38 | |
| GO:0005768 | Endosome | 8.24 × 10−38 | |
| GO:0030659 | Cytoplasmic vesicle membrane | 1.80 × 10−34 | |
| Cluster 3 | Biological Process (Gene Ontology) | ||
| GO:0016192 | Vesicle-mediated transport | 4.17 × 10−59 | |
| GO:0061025 | Membrane fusion | 6.79 × 10−59 | |
| GO:0006906 | Vesicle fusion | 1.63 × 10−53 | |
| GO:0051649 | Establishment of localization in cell | 5.79 × 10−46 | |
| GO:0016050 | Vesicle organization | 7.23 × 10−44 | |
| Molecular Function (Gene Ontology) | |||
| GO:0000149 | SNARE binding | 1.15 × 10−72 | |
| GO:0005484 | SNAP receptor activity | 5.11 × 10−70 | |
| GO:0030674 | Protein-macromolecule adaptor activity | 6.13 × 10−46 | |
| GO:0019905 | Syntaxin binding | 4.45 × 10−43 | |
| GO:0017075 | Syntaxin-1 binding | 7.64 × 10−16 | |
| Cellular Component (Gene Ontology) | |||
| GO:0031201 | SNARE complex | 2.90 × 10−91 | |
| GO:0030133 | Transport vesicle | 4.17 × 10−43 | |
| GO:0070382 | Exocytic vesicle | 3.76 × 10−41 | |
| GO:0008021 | Synaptic vesicle | 2.50 × 10−40 | |
| GO:0098796 | Membrane protein complex | 4.14 × 10−40 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, H.N.; Uversky, V.N. Intrinsic Disorder in the Host Proteins Entrapped in Rabies Virus Particles. Viruses 2024, 16, 916. https://doi.org/10.3390/v16060916
Ashraf HN, Uversky VN. Intrinsic Disorder in the Host Proteins Entrapped in Rabies Virus Particles. Viruses. 2024; 16(6):916. https://doi.org/10.3390/v16060916
Chicago/Turabian StyleAshraf, Hafiza Nimra, and Vladimir N. Uversky. 2024. "Intrinsic Disorder in the Host Proteins Entrapped in Rabies Virus Particles" Viruses 16, no. 6: 916. https://doi.org/10.3390/v16060916