

Phylogenetic and Evolutionary Analysis of Porcine Epidemic Diarrhea Virus in Guangxi Province, China, during 2020 and 2024

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Amplification and Sequencing of Targeted Genes
2.3. Sequence Comparison and Phylogenetic Analysis
2.4. Bayesian Temporal Dynamics Analysis
2.5. Recombination Events Analysis
2.6. Amino Acid Difference Analysis of PEDV S1 Gene
3. Results
3.1. Test Results of Clinical Samples
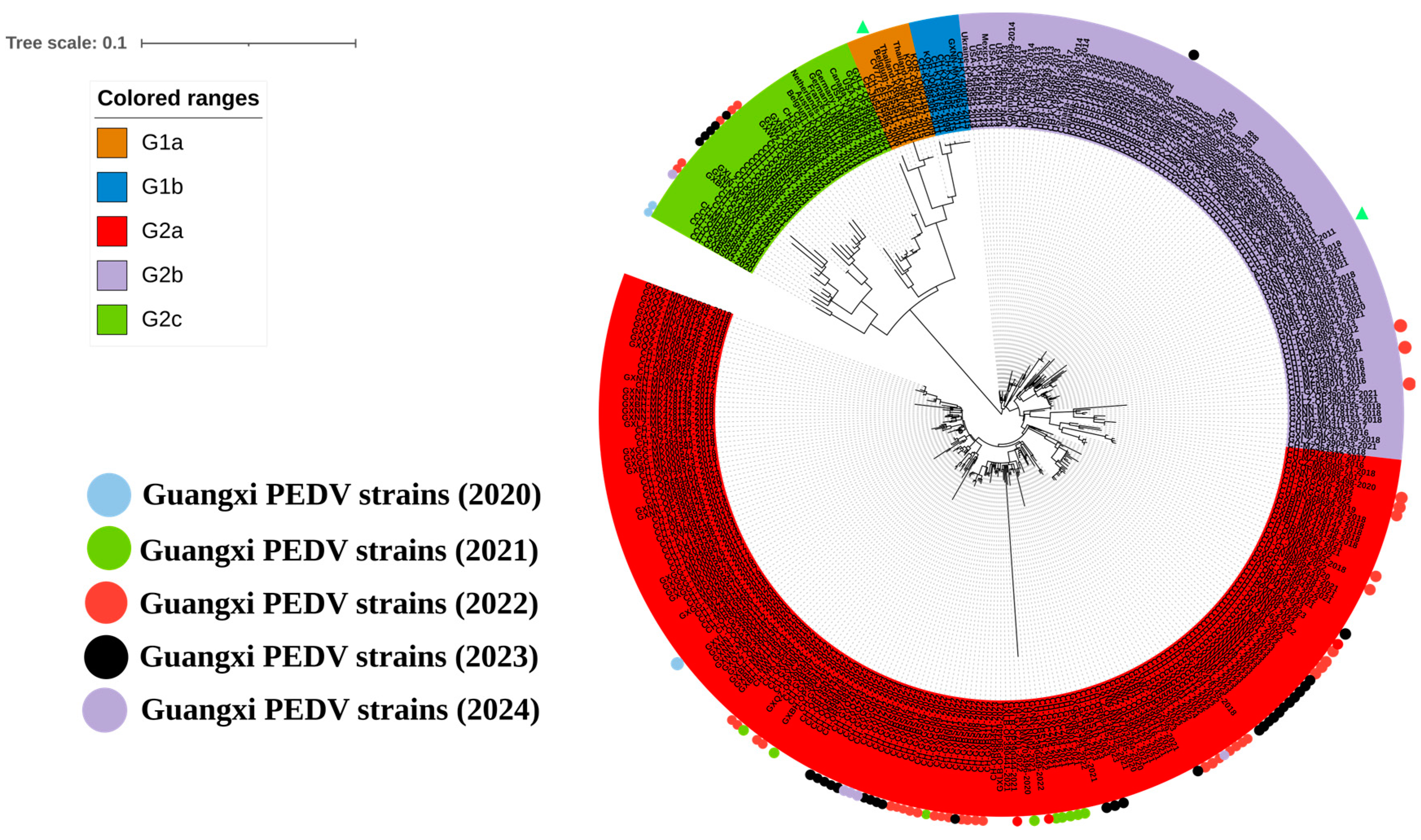
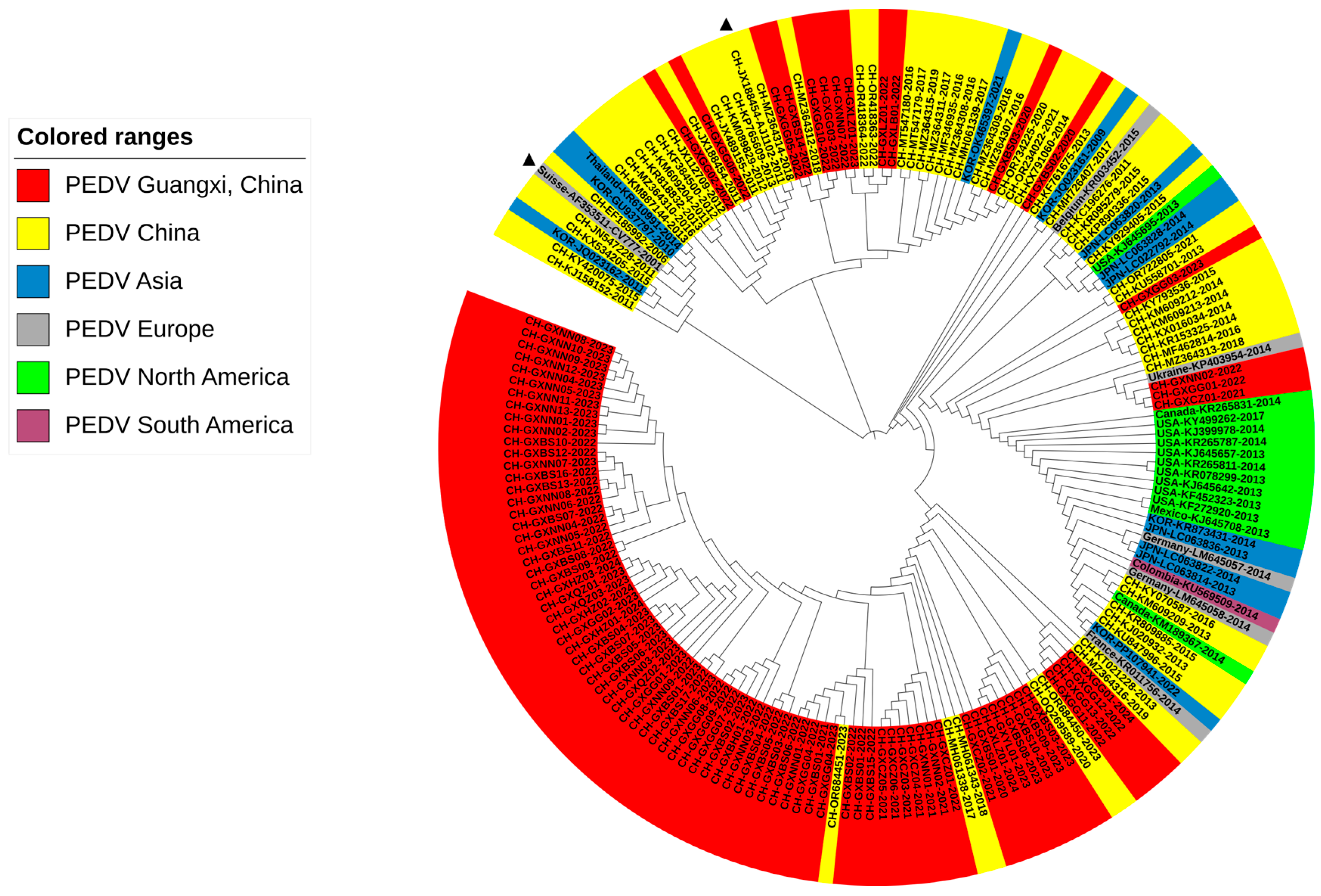
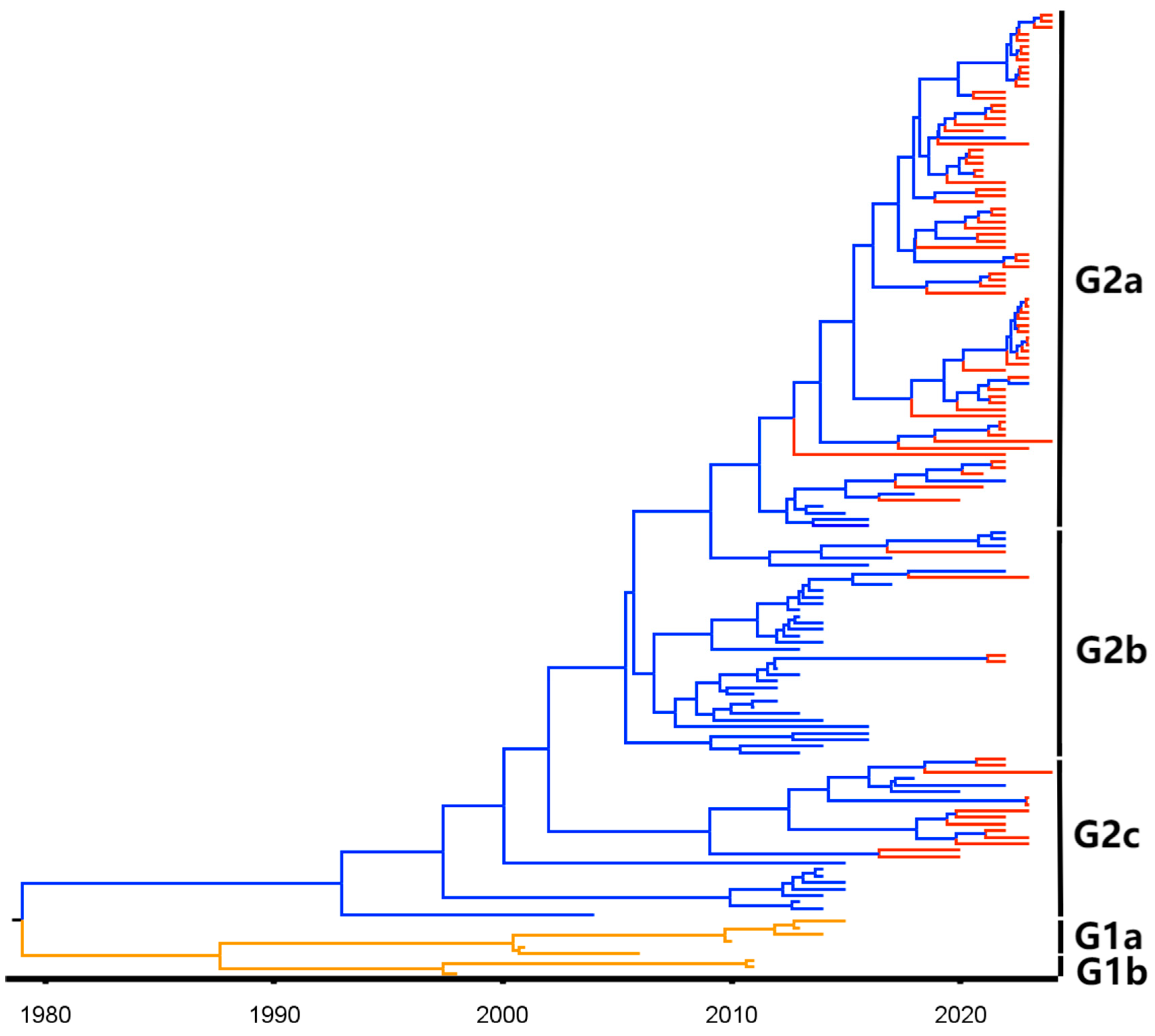
3.2. Phylogenetic Analysis Based on S1 Gene Sequences
3.3. Phylogenetic Analysis Based on S1 Gene Sequences from Guangxi Province
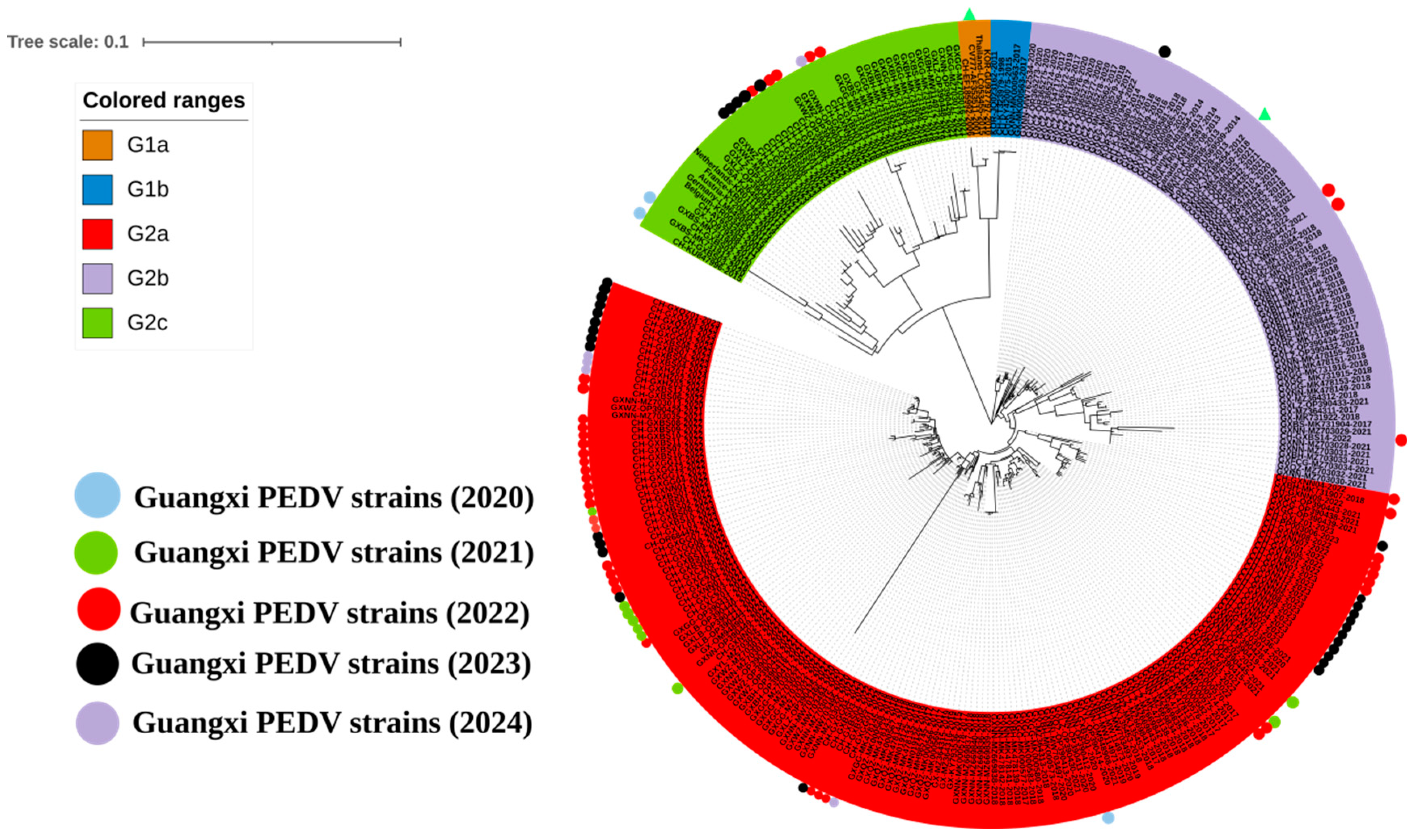
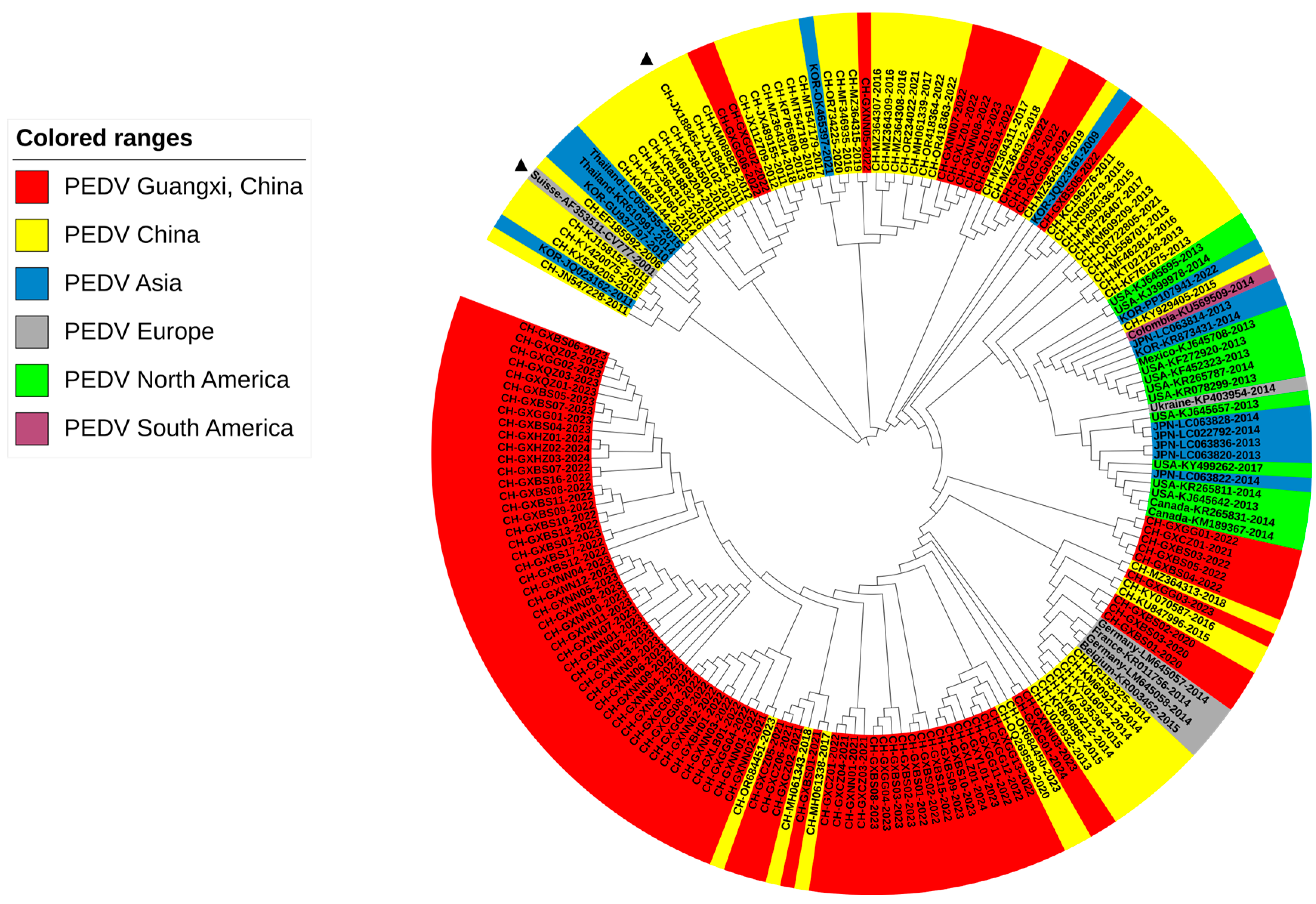
3.4. Phylogenetic Analysis Based on M Gene Sequences
3.5. Phylogenetic Analysis Based on N Gene Sequences
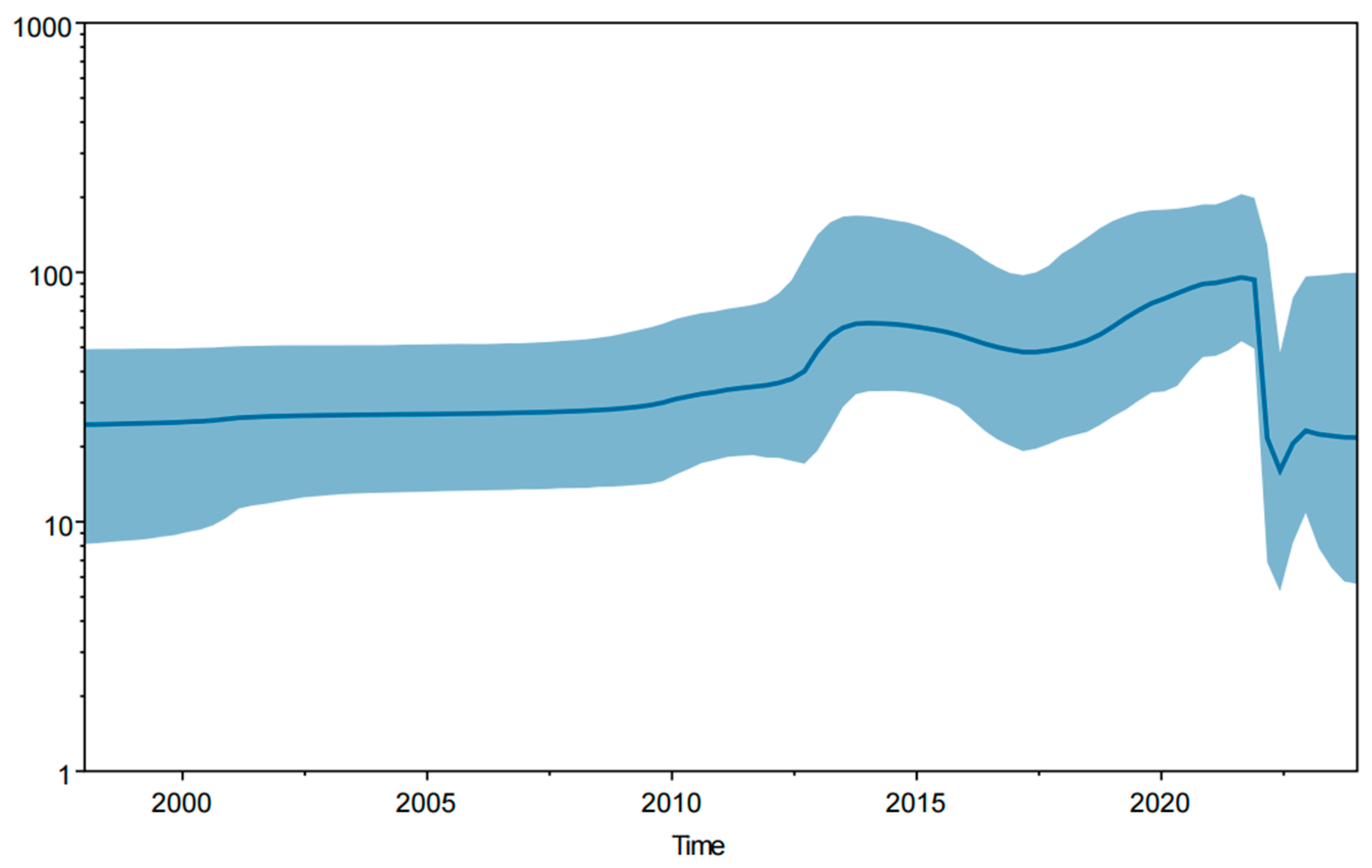
3.6. Bayesian Temporal Dynamics Analysis
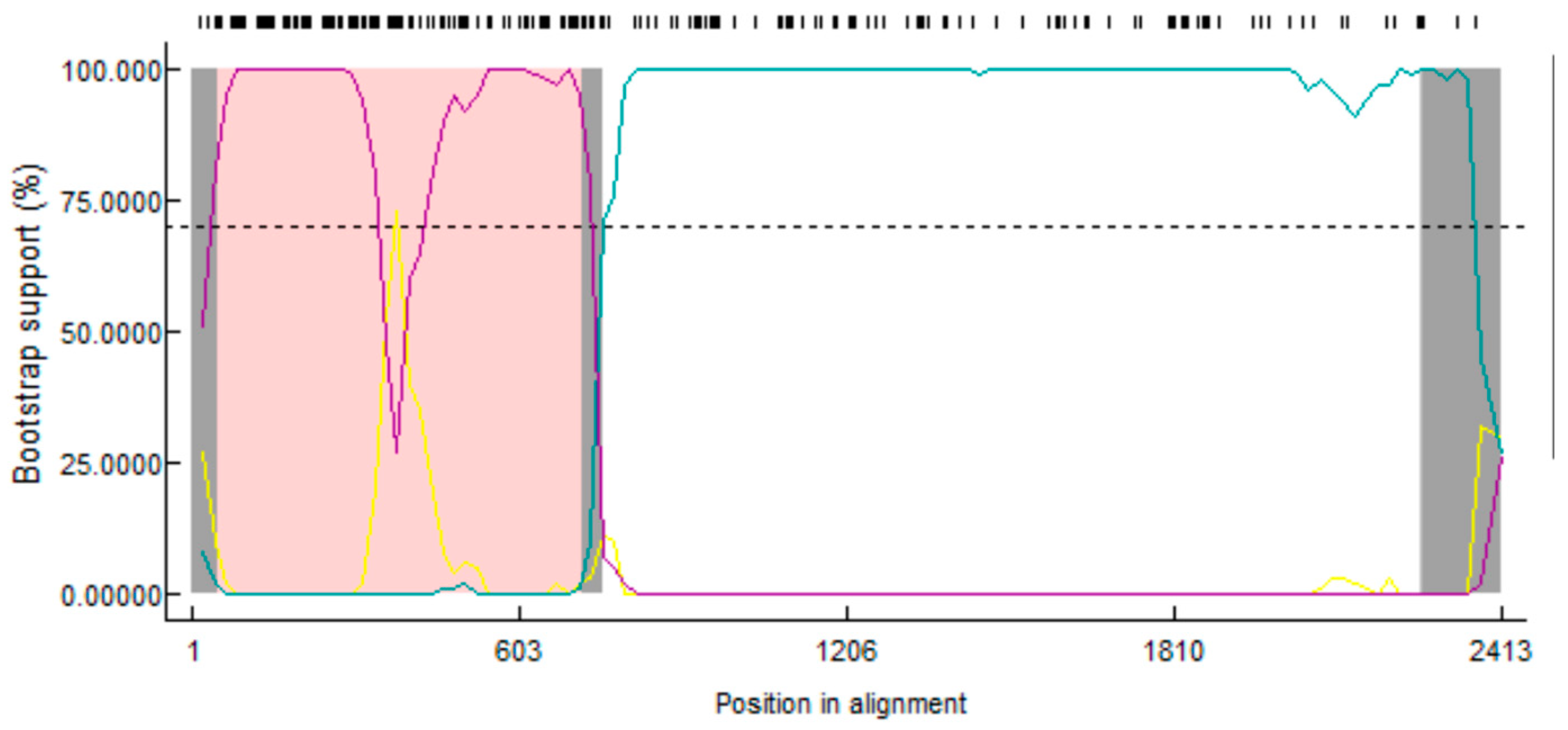
3.7. Recombination Analysis of the PEDV S1 Gene
3.8. Genetic Evolution of PEDV S1, M, and N Genes
3.9. Amino Acid Sequence Analysis of the S1 Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Chen, Y.; Zhou, J.; Wang, X.; Ma, L.; Li, J.; Yang, L.; Yuan, H.; Pang, D.; Ouyang, H. Porcine epidemic diarrhea virus: An updated overview of virus epidemiology, virulence variation patterns and virus-host interactions. Viruses 2022, 14, 2434. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; de Groot, R.J.; Haagmans, B.; Lau, S.K.P.; Neuman, B.W.; Perlman, S.; Sola, I.; van der Hoek, L.; Wong, A.C.P.; Yeh, S.H. ICTV virus taxonomy profile: Coronaviridae 2023. J. Gen. Virol. 2023, 104, 001843. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Zhang, H.; Li, L.; Yang, Y.; Zou, X.; Chen, J.; Tang, X. PEDV: Insights and advances into types, function, structure, and receptor recognition. Viruses 2022, 14, 1744. [Google Scholar] [CrossRef] [PubMed]
- Pensaert, M.B.; de Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, H.; Liu, Y.; Pan, Y.; Deng, F.; Song, Y.; Tang, X.; He, Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012, 18, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.W.; Dickerman, A.W.; Piñeyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio. 2013, 4, e00737-13. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ma, Z.; Li, Y.; Gao, S.; Xiao, S. Porcine epidemic diarrhea virus: Molecular mechanisms of attenuation and vaccines. Microb. Pathog. 2020, 149, 104553. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Jia, H.; Xiao, Q.; Fang, L.; Wang, Q. Prevention and control of swine enteric coronaviruses in China: A review of vaccine development and application. Vaccines 2023, 12, 11. [Google Scholar] [CrossRef]
- Jung, K.; Saif, L.J.; Wang, Q. Porcine epidemic diarrhea virus (PEDV): An update on etiology, transmission, pathogenesis, and prevention and control. Virus Res. 2020, 286, 198045. [Google Scholar] [CrossRef]
- Li, M.; Pan, Y.; Xi, Y.; Wang, M.; Zeng, Q. Insights and progress on epidemic characteristics, genotyping, and preventive measures of PEDV in China: A review. Microb. Pathog. 2023, 181, 106185. [Google Scholar] [CrossRef]
- Antas, M.; Woźniakowski, G. Current status of porcine epidemic diarrhoea (PED) in European pigs. J. Vet. Res. 2019, 63, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Guo, D.; Li, C.; Wei, S.; Wang, Z.; Liu, Q.; Zhang, B.; Kong, F.; Feng, L.; Sun, D. Molecular characterization of the ORF3 and S1 genes of porcine epidemic diarrhea virus Non S-INDEL strains in seven regions of China, 2015. PLoS ONE 2016, 11, e0160561. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Huang, W.; Zhong, L.; Qin, Y.; Liu, X.; Yang, C.; Wang, R.; Su, X.; Du, C.; Mi, X.; et al. Comparative characterization and pathogenicity of a novel porcine epidemic diarrhea virus (PEDV) with a naturally occurring truncated ORF3 gene coinfected with PEDVs possessing an intact ORF3 gene in piglets. Viruses 2021, 13, 1562. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Wang, Y.; Li, R.; Wang, S.; Ding, P.; Zhang, G. Accurate location of two conserved linear epitopes of PEDV utilizing monoclonal antibodies induced by S1 protein nanoparticles. Int. J. Biol. Macromol. 2023, 253, 127276. [Google Scholar] [CrossRef] [PubMed]
- Thavorasak, T.; Chulanetra, M.; Glab-Ampai, K.; Teeranitayatarn, K.; Songserm, T.; Yodsheewan, R.; Sae-Lim, N.; Lekcharoensuk, P.; Sookrung, N.; Chaicumpa, W. Novel neutralizing epitope of PEDV S1 protein identified by IgM monoclonal antibody. Viruses 2022, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yang, Y.; Zhao, J.; Geng, N.; Liu, K.; Zhao, Y.; Wang, F.; Liu, S.; Li, N.; Meng, F.; et al. Molecular epidemiological survey of porcine epidemic diarrhea in some areas of Shandong and genetic evolutionary analysis of S gene. Front. Vet. Sci. 2022, 9, 1015717. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Li, C.; Qi, S.; Yang, D.; Jiang, N.; Yin, B.; Guo, D.; Kong, F.; Yuan, D.; Feng, L.; et al. A molecular epidemiological investigation of PEDV in China: Characterization of co-infection and genetic diversity of S1-based genes. Transbound. Emerg. Dis. 2020, 67, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enríquez, A.; Herrera-Camacho, I.; Millán-Pérez-Peña, L.; Reyes-Leyva, J.; Santos-López, G.; Rivera-Benítez, J.F.; Rosas-Murrieta, N.H. Predicted 3D model of the M protein of porcine epidemic diarrhea virus and analysis of its immunogenic potential. PLoS ONE 2022, 17, e0263582. [Google Scholar] [CrossRef]
- Zhou, J.; Qiu, Y.; Zhao, J.; Wang, Y.; Zhu, N.; Wang, D.; Cui, Y.; Guo, J.; Sun, T.; Ji, Y.; et al. The network of interactions between the porcine epidemic diarrhea virus nucleocapsid and host cellular proteins. Viruses 2022, 14, 2269. [Google Scholar] [CrossRef]
- Bai, J.; Du, C.; Lu, Y.; Wang, R.; Su, X.; Yu, K.; Qin, Q.; Chen, Y.; Wei, Z.; Huang, W.; et al. Phylogenetic and spatiotemporal analyses of porcine epidemic diarrhea virus in Guangxi, China during 2017–2022. Animals 2023, 13, 1215. [Google Scholar] [CrossRef]
- Tian, Y.; Yang, X.; Li, H.; Ma, B.; Guan, R.; Yang, J.; Chen, D.; Han, X.; Zhou, L.; Song, Z.; et al. Molecular characterization of porcine epidemic diarrhea virus associated with outbreaks in southwest China during 2014–2018. Transbound. Emerg. Dis. 2021, 68, 3482–3497. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Miao, Y.; Bi, W.; Xiang, C.; Li, W.; Zhang, R.; Li, Q.; Yang, Z. Porcine epidemic diarrhea virus: Etiology, epidemiology, antigenicity, and control strategies in China. Animals 2024, 14, 294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Shi, K.; Long, F.; Zhao, K.; Feng, S.; Yin, Y.; Xiong, C.; Qu, S.; Lu, W.; Li, Z. A quadruplex qRT-PCR for differential detection of four porcine enteric coronaviruses. Vet. Sci. 2022, 9, 634. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gao, Y.; Ma, Y.; Shi, K.; Shi, Y.; Feng, S.; Yin, Y.; Long, F.; Sun, W. Genetic and evolutionary analysis of porcine deltacoronavirus in Guangxi province, southern China, from 2020 to 2023. Microorganisms. 2024, 12, 416. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Chen, L.; Song, J.; Liu, H.; Cai, J.; Lin, Q.; Xu, C.; Ding, C.; Liao, M.; Ren, T.; Xiang, B. Phylodynamic analyses of class I Newcastle disease virus isolated in China. Transbound. Emerg. Dis. 2021, 68, 1294–1304. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Kocherhans, R.; Bridgen, A.; Ackermann, M.; Tobler, K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes 2001, 23, 137–144. [Google Scholar] [CrossRef]
- Bi, J.; Zeng, S.; Xiao, S.; Chen, H.; Fang, L. Complete genome sequence of porcine epidemic diarrhea virus strain AJ1102 isolated from a suckling piglet with acute diarrhea in China. J. Virol. 2012, 86, 10910–10911. [Google Scholar] [CrossRef]
- Turlewicz-Podbielska, H.; Pomorska-Mól, M. Porcine coronaviruses: Overview of the state of the art. Virol. Sin. 2021, 36, 833–851. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Jiao, D.; Zhang, R.; Zhou, J.; Guo, R.; Yu, Z.; Shi, D.; Zhao, Y.; Gu, J.; Niu, B.; et al. Origin and epidemic status of porcine epidemic diarrhea virus variants in China. Transbound. Emerg. Dis. 2020, 67, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Hao, H.; Li, M.; Niu, J.; Hu, Y.; Zhao, X.; Li, Q. Expression and purification of a PEDV-neutralizing antibody and its functional verification. Viruses 2021, 13, 472. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, X.; Wei, S.; Chen, J.; Feng, L. Epidemiology and vaccine of porcine epidemic diarrhea virus in China: A mini-review. J. Vet. Med. Sci. 2016, 78, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Yuan, W.; Peng, Q.; Zhang, F.; Ye, Y.; Huang, D.; Ding, Z.; Lin, L.; He, H.; et al. Evaluation of cross-protection between G1a- and G2a-genotype porcine epidemic diarrhea viruses in suckling piglets. Animals 2020, 10, 1674. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Qi, W.; Yang, Y.; Liu, Z.; An, T.; Wu, X.; Chen, J. Prevention and control strategies of African swine fever and progress on pig farm repopulation in China. Viruses 2021, 13, 2552. [Google Scholar] [CrossRef] [PubMed]
- Juszkiewicz, M.; Walczak, M.; Woźniakowski, G.; Podgórska, K. African swine fever: Transmission, spread, and control through biosecurity and disinfection, including Polish trends. Viruses 2023, 15, 2275. [Google Scholar] [CrossRef]
- Tian, L.; Luo, Y.; Wen, T.; Yang, W.; Zhao, Y.; Huang, P.; He, H.; Wu, J.; Li, Z.; Pan, C. A quadruple protection procedure for resuming pig production in small-scale ASFV-positive farms in China. Curr. Res. Microb. Sci. 2020, 2, 100014. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Fang, L.; Ye, X.; Chen, J.; Xu, S.; Zhu, X.; Miao, Y.; Wang, D.; Xiao, S. Evolutionary and genotypic analyses of global porcine epidemic diarrhea virus strains. Transbound. Emerg. Dis. 2019, 66, 111–118. [Google Scholar] [CrossRef]
- Wen, F.; Yang, J.; Li, A.; Gong, Z.; Yang, L.; Cheng, Q.; Wang, C.; Zhao, M.; Yuan, S.; Chen, Y.; et al. Genetic characterization and phylogenetic analysis of porcine epidemic diarrhea virus in Guangdong, China, between 2018 and 2019. PLoS ONE 2021, 16, e0253622. [Google Scholar] [CrossRef]
- López-Figueroa, C.; Cano, E.; Navarro, N.; Pérez-Maíllo, M.; Pujols, J.; Núñez, J.I.; Vergara-Alert, J.; Segalés, J. Clinical, pathological and virological outcomes of tissue-homogenate-derived and cell-adapted strains of porcine epidemic diarrhea virus (PEDV) in a neonatal pig model. Viruses 2023, 16, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fan, B.; Song, X.; Gao, J.; Guo, R.; Yi, C.; He, Z.; Hu, H.; Jiang, J.; Zhao, L.; et al. PEDV-spike-protein-expressing mRNA vaccine protects piglets against PEDV challenge. mBio. 2024, 15, e0295823. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Xiao, S. Porcine epidemic diarrhea in China. Virus Res. 2016, 226, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Su, X.; Du, C.; Mo, L.; Ke, P.; Wang, R.; Zhong, L.; Yang, C.; Chen, Y.; Wei, Z.; et al. Genetic diversity of porcine epidemic diarrhea virus with a naturally occurring truncated ORF3 gene found in Guangxi, China. Front. Vet. Sci. 2020, 7, 435. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Liu, X.; Lu, Y.; Long, M.; Bai, J.; Qin, Q.; Su, X.; He, G.; Mi, X.; Yang, C.; et al. Biological characteristics and pathogenicity analysis of a low virulence G2a porcine epidemic diarrhea virus. Microbiol. Spectr. 2023, 11, e0453522. [Google Scholar] [CrossRef] [PubMed]
- Salamat, S.E.A.; Collantes, T.M.A.; Lumbera, W.M.L.; Tablizo, F.A.; Mutia, C.T.M.; Ong, J.D.P.; Bandoy, D.J.D.R. Sequence analysis of new variants of porcine epidemic diarrhea virus in Luzon, Philippines, in 2017. Arch. Virol. 2021, 166, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Li, C.; Qiu, Y.; Qi, W.; Qiu, M.; Li, J.; Lin, H.; Zheng, W.; Zhu, J.; Chen, N. Genomic characterizations of porcine epidemic diarrhea viruses (PEDV) in diarrheic piglets and clinically healthy adult pigs from 2019 to 2022 in China. Animals 2023, 13, 1562. [Google Scholar] [CrossRef]
- Lin, C.M.; Saif, L.J.; Marthaler, D.; Wang, Q. Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Res. 2016, 226, 20–39. [Google Scholar] [CrossRef]
- Rivera-Benítez, J.F.; Martínez-Bautista, R.; González-Martínez, R.; De la Luz-Armendáriz, J.; Herrera-Camacho, I.; Rosas-Murrieta, N.; Márquez-Valdelamar, L.; Lara, R. Phylogenetic and molecular analysis of the porcine epidemic diarrhea virus in Mexico during the first reported outbreaks (2013–2017). Viruses 2024, 16, 309. [Google Scholar] [CrossRef]
- Sato, T.; Takeyama, N.; Katsumata, A.; Tuchiya, K.; Kodama, T.; Kusanagi, K. Mutations in the spike gene of porcine epidemic diarrhea virus associated with growth adaptation in vitro and attenuation of virulence in vivo. Virus Genes 2011, 43, 72–78. [Google Scholar] [CrossRef]
- Lee, C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol. J. 2015, 12, 193. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y.; Liu, Y.; Chen, Y.; Jiao, W.; Feng, H.; Wei, Q.; Wang, J.; Zhang, Y.; Zhang, G. Isolation and identification of a recombinant porcine epidemic diarrhea virus with a novel insertion in S1 domain. Front. Microbiol. 2021, 12, 667084. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence (5′→3′) | Product/bp |
|---|---|---|---|
| S1 | PEDV-SI-F PEDV-SI-R PEDV-SII-F PEDV-SII-R PEDV-SIII-F PEDV-SIII-R | CCATTAGTGATGTTGTGTTAG GCACAGCAGCTCCATT CCACATACCAGAAGGTTTTAG CCAGTAATCAACTCACCCTT CCTGAGTTTGGTAGTGGTGTTA GGTGAGTAATTGTTTACAACGAGAG | 1040 1146 654 |
| M | PEDV-M-F PEDV-M-R | GTCTTACATGCGAATTGACC GGCATAGAGAGATAATGGCA | 808 |
| N | PEDV-N-F PEDV-N-R | TGCGGTTCTCACAGATAGTG AAGTCGCTAGAAAAACACTCAGTAAT | 1462 |
| Guangxi Strain (n = 92) | Percentage of Nucleotide (Amino Acid) Identity (%) | ||
|---|---|---|---|
| CV777 | AJ1102 | ||
| S1 | G2a (n = 75) | 91.1–92.0% (89.4–90.9%) | 96.9–98.8% (96.5–98.1%) |
| G2b (n = 4) | 91.2–92.0% (89.1–90.9%) | 96.3–99.2% (95.6–99.1%) | |
| G2c (n = 13) | 93.6–94.3% (92.4–94.0%) | 92.0–92.6% (90.5–92.1%) | |
| M | 96.6–98.1% (93.6–99.4%) | 97.1–99.6% (93.6–99.4%) | |
| N | 94.8–95.8% (80.2–93.8%) | 95.3–99.7% (82.7–95.6%) | |
| Guangxi Strain (n = 92) | Percentage of Nucleotide (Amino Acid) Identity (%) | ||
|---|---|---|---|
| The Strains Obtained in This Study | The Other Strains from Guangxi Province | The Other Strains from China and Other Countries | |
| S1 | 91.8–99.9% (89.3–99.5%) | 91.2–99.9% (89.0–99.5%) | 90.8–99.8% (88.3–99.4%) |
| M | 97.0–100.0% (96.2–100.0%) | 96.6–99.9% (92.3–100.0%) | 95.1–99.9% (84.3–100.0%) |
| N | 95.3–99.9% (82.6–100.0%) | 95.1–99.9% (77.3–100.0%) | 93.8–99.7% (73.3–100.0%) |
| Year | G1b | G2a | G2b | G2c | Total |
|---|---|---|---|---|---|
| 2011 | 0 | 0 | 0 | 1 | 1 |
| 2015 | 0 | 1 | 0 | 0 | 1 |
| 2016 | 0 | 0 | 4 | 0 | 4 |
| 2017 | 2 | 11 | 8 | 1 | 22 |
| 2018 | 0 | 27 | 26 | 11 | 64 |
| 2019 | 0 | 5 | 1 | 3 | 9 |
| 2020 | 0 | 13 | 10 | 2 | 25 |
| 2021 | 0 | 39 | 17 | 8 | 64 |
| 2022 | 0 | 35 | 4 | 8 | 47 |
| 2023 | 0 | 26 | 1 | 5 | 32 |
| 2024 | 0 | 4 | 0 | 1 | 5 |
| Total | 2 | 161 | 71 | 40 | 274 |
| Gene | Evolutionary Rate (S/S/Y) | 95% HPD Interval |
|---|---|---|
| S1 | 1.527 × 10−3 | 1.2014 × 10−3–1.8418 × 10−3 |
| M | 1.518 × 10−4 | 1.269 × 10−4–1.7983 × 10−4 |
| N | 1.076 × 10−3 | 8.2589 × 10−4–1.3442 × 10−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, K.; Li, B.; Shi, Y.; Feng, S.; Yin, Y.; Long, F.; Pan, Y.; Wei, Y. Phylogenetic and Evolutionary Analysis of Porcine Epidemic Diarrhea Virus in Guangxi Province, China, during 2020 and 2024. Viruses 2024, 16, 1126. https://doi.org/10.3390/v16071126
Shi K, Li B, Shi Y, Feng S, Yin Y, Long F, Pan Y, Wei Y. Phylogenetic and Evolutionary Analysis of Porcine Epidemic Diarrhea Virus in Guangxi Province, China, during 2020 and 2024. Viruses. 2024; 16(7):1126. https://doi.org/10.3390/v16071126
Chicago/Turabian StyleShi, Kaichuang, Biao Li, Yuwen Shi, Shuping Feng, Yanwen Yin, Feng Long, Yi Pan, and Yingyi Wei. 2024. "Phylogenetic and Evolutionary Analysis of Porcine Epidemic Diarrhea Virus in Guangxi Province, China, during 2020 and 2024" Viruses 16, no. 7: 1126. https://doi.org/10.3390/v16071126
APA StyleShi, K., Li, B., Shi, Y., Feng, S., Yin, Y., Long, F., Pan, Y., & Wei, Y. (2024). Phylogenetic and Evolutionary Analysis of Porcine Epidemic Diarrhea Virus in Guangxi Province, China, during 2020 and 2024. Viruses, 16(7), 1126. https://doi.org/10.3390/v16071126