

Whole-Genome Characterization of Rotavirus G9P[6] and G9P[4] Strains That Emerged after Rotavirus Vaccine Introduction in Mozambique

 , , , , ,
, , , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Viral Genomic dsRNA Extraction, cDNA Library Building and Illumina MiSeq Sequencing
2.3. Data Analysis
2.3.1. Genome Assembly
2.3.2. Determination of RVA Genotypes
2.3.3. Phylogenetic Analysis
2.3.4. Nucleotide Sequence Accession Numbers
3. Results
3.1. Genome Constellations
3.2. Phylogenetic Analysis
3.2.1. VP7 Encoding Gene
G9 Genotype
G3, G2 and G1 Genotypes
3.2.2. VP4 Encoding Gene
P[4], P[6] and P[8] Genotypes
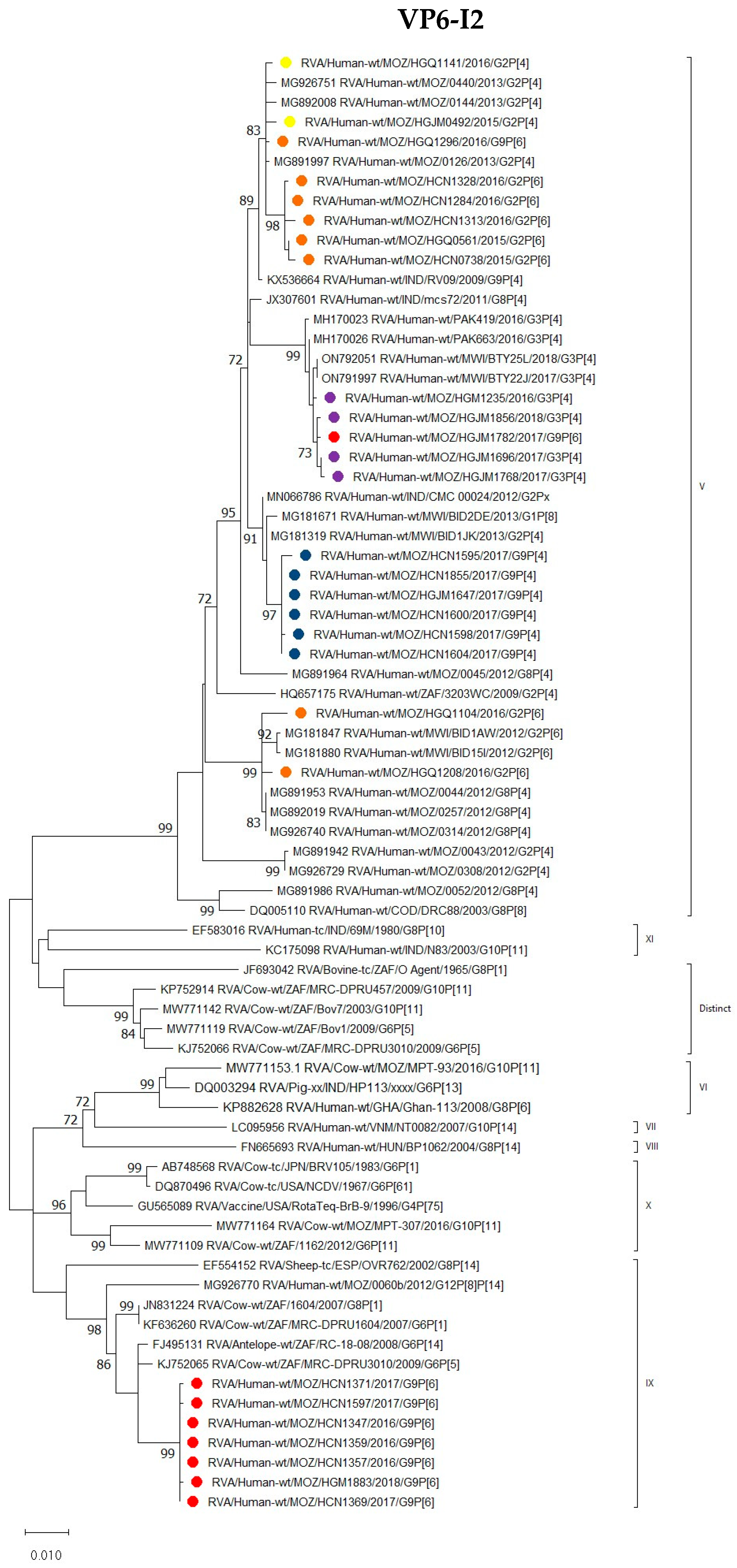
3.2.3. VP1–VP3 and VP6 Encoding Genes
VP1–VP3
VP6
3.2.4. NSP1-NSP5/NSP6 Encoding Genes
3.3. Mvista Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tate, J.E.; Burton, A.H.; Boschi-Pinto, C.; Parashar, U.D. Global, Regional, and National Estimates of Rotavirus Mortality in Children <5 Years of Age, 2000–2013. Clin. Infect. Dis. 2016, 62, S96–S105. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Ramani, S.; Tate, J.E.; Parashar, U.D.; Svensson, L.; Hagbom, M.; Franco, M.A.; Greenberg, H.B.; O’Ryan, M.; Kang, G.; et al. Rotavirus Infection. Nat. Rev. Dis. Primer 2017, 3, 17083. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.; Blacker, B.F.; Khalil, I.A.; Rao, P.C.; Cao, S.; Zimsen, S.R.; Albertson, S.B.; Stanaway, J.D.; Deshpande, A.; Abebe, Z.; et al. Estimates of the Global, Regional, and National Morbidity, Mortality, and Aetiologies of Diarrhoea in 195 Countries: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1211–1228. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Attoui, H.; Bányai, K.; Brussaard, C.P.D.; Danthi, P.; del Vas, M.; Dermody, T.S.; Duncan, R.; Fāng, Q.; Johne, R.; et al. ICTV Virus Taxonomy Profile: Sedoreoviridae 2022. J. Gen. Virol. 2022, 103, 001782. [Google Scholar] [CrossRef] [PubMed]
- Bányai, K.; Estes, M.K.; Martella, V.; Parashar, U.D. Viral Gastroenteritis. Lancet 2018, 392, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Rotavirus Classification Working Group: RCWG. Available online: https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/rcwg (accessed on 27 July 2023).
- Mijatovic-Rustempasic, S.; Jaimes, J.; Perkins, C.; Ward, M.L.; Esona, M.D.; Gautam, R.; Lewis, J.; Sturgeon, M.; Panjwani, J.; Bloom, G.A.; et al. Rotavirus Strain Trends in United States, 2009–2016: Results from the National Rotavirus Strain Surveillance System (NRSSS). Viruses 2022, 14, 1775. [Google Scholar] [CrossRef]
- Burke, R.M.; Tate, J.E.; Kirkwood, C.D.; Steele, A.D.; Parashar, U.D. Current and New Rotavirus Vaccines. Curr. Opin. Infect. Dis. 2019, 32, 435–444. [Google Scholar] [CrossRef]
- Seheri, L.M.; Magagula, N.B.; Peenze, I.; Rakau, K.; Ndadza, A.; Mwenda, J.M.; Weldegebriel, G.; Steele, A.D.; Mphahlele, M.J. Rotavirus Strain Diversity in Eastern and Southern African Countries before and after Vaccine Introduction. Vaccine 2018, 36, 7222–7230. [Google Scholar] [CrossRef]
- Antoni, S.; Nakamura, T.; Cohen, A.L.; Mwenda, J.M.; Weldegebriel, G.; Biey, J.N.M.; Shaba, K.; Rey-Benito, G.; de Oliveira, L.H.; da Costa Oliveira, M.T.; et al. Rotavirus Genotypes in Children under Five Years Hospitalized with Diarrhea in Low and Middle-Income Countries: Results from the WHO-Coordinated Global Rotavirus Surveillance Network. PLoS Glob. Public Health 2023, 3, e0001358. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of Rotavirus Strain Nomenclature Proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Van Ranst, M. Genotype Constellation and Evolution of Group A Rotaviruses Infecting Humans. Curr. Opin. Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gómara, M.; Maes, P.; Patton, J.T.; et al. Full Genome-Based Classification of Rotaviruses Reveals a Common Origin between Human Wa-Like and Porcine Rotavirus Strains and Human DS-1-Like and Bovine Rotavirus Strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef]
- de Deus, N.; Chilaúle, J.J.; Cassocera, M.; Bambo, M.; Langa, J.S.; Sitoe, E.; Chissaque, A.; Anapakala, E.; Sambo, J.; Guimarães, E.L.; et al. Early Impact of Rotavirus Vaccination in Children Less than Five Years of Age in Mozambique. Vaccine 2018, 36, 7205–7209. [Google Scholar] [CrossRef]
- João, E.D.; Munlela, B.; Chissaque, A.; Chilaúle, J.; Langa, J.; Augusto, O.; Boene, S.S.; Anapakala, E.; Sambo, J.; Guimarães, E.; et al. Molecular Epidemiology of Rotavirus A Strains Pre- and Post-Vaccine (Rotarix®) Introduction in Mozambique, 2012–2019: Emergence of Genotypes G3P[4] and G3P[8]. Pathogens 2020, 9, 671. [Google Scholar] [CrossRef]
- Chissaque, A.; Burke, R.M.; Guimarães, E.L.; Manjate, F.; Nhacolo, A.; Chilaúle, J.; Munlela, B.; Chirinda, P.; Langa, J.S.; Cossa-Moiane, I.; et al. Effectiveness of Monovalent Rotavirus Vaccine in Mozambique, a Country with a High Burden of Chronic Malnutrition. Vaccines 2022, 10, 449. [Google Scholar] [CrossRef] [PubMed]
- Manjate, F.; João, E.D.; Chirinda, P.; Garrine, M.; Vubil, D.; Nobela, N.; Kotloff, K.; Nataro, J.P.; Nhampossa, T.; Acácio, S.; et al. Molecular Epidemiology of Rotavirus Strains in Symptomatic and Asymptomatic Children in Manhiça District, Southern Mozambique 2008–2019. Viruses 2022, 14, 134. [Google Scholar] [CrossRef]
- Strydom, A.; Motanyane, L.; Nyaga, M.M.; João, E.D.; Cuamba, A.; Mandomando, I.; Cassocera, M.; de Deus, N.; O’Neill, H. Whole-Genome Characterization of G12 Rotavirus Strains Detected in Mozambique Reveals a Co-Infection with a GXP[14] Strain of Possible Animal Origin. J. Gen. Virol. 2019, 100, 932–937. [Google Scholar] [CrossRef]
- Strydom, A.; João, E.D.; Motanyane, L.; Nyaga, M.M.; Christiaan Potgieter, A.; Cuamba, A.; Mandomando, I.; Cassocera, M.; de Deus, N.; O’Neill, H.G. Whole Genome Analyses of DS-1-like Rotavirus A Strains Detected in Children with Acute Diarrhoea in Southern Mozambique Suggest Several Reassortment Events. Infect. Genet. Evol. 2019, 69, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Munlela, B.; João, E.D.; Donato, C.M.; Strydom, A.; Boene, S.S.; Chissaque, A.; Bauhofer, A.F.L.; Langa, J.; Cassocera, M.; Cossa-Moiane, I.; et al. Whole Genome Characterization and Evolutionary Analysis of G1P[8] Rotavirus A Strains during the Pre- and Post-Vaccine Periods in Mozambique (2012–2017). Pathogens 2020, 9, 1026. [Google Scholar] [CrossRef] [PubMed]
- Gentsch, J.R.; Glass, R.I.; Woods, P.; Gouvea, V.; Gorziglia, M.; Flores, J.; Das, B.K.; Bhan, M.K. Identification of Group A Rotavirus Gene 4 Types by Polymerase Chain Reaction. J. Clin. Microbiol. 1992, 30, 1365–1373. [Google Scholar] [CrossRef]
- Gouvea, V.; Glass, R.I.; Woods, P.; Taniguchi, K.; Clark, H.F.; Forrester, B.; Fang, Z.Y. Polymerase Chain Reaction Amplification and Typing of Rotavirus Nucleic Acid from Stool Specimens. J. Clin. Microbiol. 1990, 28, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Iturriza-Gómara, M.; Kang, G.; Gray, J. Rotavirus Genotyping: Keeping up with an Evolving Population of Human Rotaviruses. J. Clin. Virol. 2004, 31, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Heylen, E.; Damanka, S.; Pietsch, C.; Donato, C.; Tamura, T.; Kulkarni, R.; Arora, R.; Cunliffe, N.; Maunula, L.; et al. Emerging OP354-Like P[8] Rotaviruses Have Rapidly Dispersed from Asia to Other Continents. Mol. Biol. Evol. 2015, 32, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Heylen, E.; Tamim, S.; McAllen, J.K.; Kirkness, E.F.; Akopov, A.; De Coster, S.; Van Ranst, M.; Matthijnssens, J. Comparative Analysis of the RotarixTM Vaccine Strain and G1P[8] Rotaviruses Detected before and after Vaccine Introduction in Belgium. PeerJ 2017, 5, e2733. [Google Scholar] [CrossRef] [PubMed]
- Damanka, S.A.; Kwofie, S.; Dennis, F.E.; Lartey, B.L.; Agbemabiese, C.A.; Doan, Y.H.; Adiku, T.K.; Katayama, K.; Enweronu-Laryea, C.C.; Armah, G.E. Whole Genome Characterization and Evolutionary Analysis of OP354-like P[8] Rotavirus A Strains Isolated from Ghanaian Children with Diarrhoea. PLoS ONE 2019, 14, e0218348. [Google Scholar] [CrossRef] [PubMed]
- Jere, K.C.; Mlera, L.; O’Neill, H.G.; Potgieter, A.C.; Page, N.A.; Seheri, M.L.; Van Dijk, A.A. Whole Genome Analyses of African G2, G8, G9, and G12 Rotavirus Strains Using Sequence-independent Amplification and 454® Pyrosequencing. J. Med. Virol. 2011, 83, 2018–2042. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, P.N.; Potgieter, R.-L.; Simwaka, J.; Mpabalwani, E.M.; Mwenda, J.M.; Mogotsi, M.T.; Magagula, N.; Esona, M.D.; Steele, A.D.; Seheri, M.L.; et al. Genomic Analysis of G2P[4] Group A Rotaviruses in Zambia Reveals Positive Selection in Amino Acid Site 7 of Viral Protein 3. Viruses 2023, 15, 501. [Google Scholar] [CrossRef]
- Agbemabiese, C.A.; Nakagomi, T.; Damanka, S.A.; Dennis, F.E.; Lartey, B.L.; Armah, G.E.; Nakagomi, O. Sub-Genotype Phylogeny of the Non-G, Non-P Genes of Genotype 2 Rotavirus A Strains. PLoS ONE 2019, 14, e0217422. [Google Scholar] [CrossRef]
- Pradhan, G.N.; Walimbe, A.M.; Chitambar, S.D. Molecular Characterization of Emerging G9P[4] Rotavirus Strains Possessing a Rare E6 NSP4 or T1 NSP3 Genotype on a Genogroup-2 Backbone Using a Refined Classification Framework. J. Gen. Virol. 2016, 97, 3139–3153. [Google Scholar] [CrossRef]
- Tatte, V.S.; Maran, D.; Walimbe, A.M.; Gopalkrishna, V. Rotavirus G9P[4], G9P[6] and G1P[6] Strains Isolated from Children with Acute Gastroenteritis in Pune, Western India, 2013–2015: Evidence for Recombination in Genes Encoding VP3, VP4 and NSP1. J. Gen. Virol. 2019, 100, 1605–1630. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.; Pachter, L.; Poliakov, A.; Rubin, E.; Dubchak, I. VISTA: Computational Tools for Comparative Genomics. Available online: https://genome.lbl.gov/vista/mvista/mvistacite.shtml (accessed on 20 October 2023).
- Doan, Y.H.; Dennis, F.E.; Takemae, N.; Haga, K.; Shimizu, H.; Appiah, M.G.; Lartey, B.L.; Damanka, S.A.; Hayashi, T.; Suzuki, T.; et al. Emergence of Intergenogroup Reassortant G9P[4] Strains Following Rotavirus Vaccine Introduction in Ghana. Viruses 2023, 15, 2453. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.P.; Kaida, A.; Ono, A.; Kubo, H.; Iritani, N. Detection and Characterization of a Human G9P[4] Rotavirus Strain in Japan. J. Med. Virol. 2015, 87, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Roy, S.; Esona, M.D.; Mijatovic-Rustempasic, S.; Hardy, C.; Wang, Y.; Cortese, M.; Bowen, M.D. Full Genome Sequence of a Reassortant Human G9P[4] Rotavirus Strain. Genome Announc. 2014, 2, e01284-14. [Google Scholar] [CrossRef] [PubMed]
- Ianiro, G.; Recanatini, C.; D’Errico, M.M.; Monini, M. Uncommon G9P[4] Group A Rotavirus Strains Causing Dehydrating Diarrhea in Young Children in Italy. Infect. Genet. Evol. 2018, 64, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Damanka, S.; Dennis, F.E.; Agbemabiese, C.; Lartey, B.; Adiku, T.; Nyarko, K.; Enweronu-Laryea, C.C.; Sagoe, K.W.; Ofori, M.; Rodrigues, O.; et al. Identification of OP354-like Human Rotavirus Strains with Subtype P[8]b in Ghanaian Children with Diarrhoea. Virol. J. 2016, 13, 69. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Potgieter, C.A.; Ciarlet, M.; Parreño, V.; Martella, V.; Bányai, K.; Garaicoechea, L.; Palombo, E.A.; Novo, L.; Zeller, M.; et al. Are Human P[14] Rotavirus Strains the Result of Interspecies Transmissions from Sheep or Other Ungulates That Belong to the Mammalian Order Artiodactyla? J. Virol. 2009, 83, 2917–2929. [Google Scholar] [CrossRef] [PubMed]
- Agbla, J.M.; Esona, M.D.; Jaimes, J.; Gautam, R.; Agbankpé, A.J.; Katz, E.; Dougnon, T.V.; Capo-Chichi, A.; Ouedraogo, N.; Razack, O.; et al. Whole Genome Analysis of Rotavirus Strains Circulating in Benin before Vaccine Introduction, 2016–2018. Virus Res. 2022, 313, 198715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, J.; Jiang, Y.-Z.; Xu, J.-Q.; Guan, X.-H.; Wang, L.-Q.; Chen, J.; Liang, Y. Genotype Distribution and Evolutionary Analysis of Rotavirus Associated with Acute Diarrhea Outpatients in Hubei, China, 2013–2016. Virol. Sin. 2022, 37, 503–512. [Google Scholar] [CrossRef]
- Khakha, S.A.; Varghese, T.; Giri, S.; Durbin, A.; Tan, G.S.; Kalaivanan, M.; Prasad, J.H.; Kang, G. Whole-Genome Characterization of Common Rotavirus Strains Circulating in Vellore, India from 2002 to 2017: Emergence of Non-Classical Genomic Constellations. Gut Pathog. 2023, 15, 44. [Google Scholar] [CrossRef]
- Doan, Y.H.; Suzuki, Y.; Fujii, Y.; Haga, K.; Fujimoto, A.; Takai-Todaka, R.; Someya, Y.; Nayak, M.K.; Mukherjee, A.; Imamura, D.; et al. Complex Reassortment Events of Unusual G9P[4] Rotavirus Strains in India between 2011 and 2013. Infect. Genet. Evol. 2017, 54, 417–428. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | VP7 | VP4 | VP6 | VP1 | VP2 | VP3 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5/6 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Segment | 9 | 4 | 6 | 1 | 2 | 3 | 5 | 8 | 7 | 10 | 11 |
| Wa-like | G1 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| DS1-like | G2 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1357/2016/G9P[6] | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1359/2016/G9P[6] | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1369/2017/G9P[6] | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1371/2017/G9P[6] | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1597/2017/G9P[6] | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HGJM1782/2017/G9P[6] | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HGQ1296/2016/G9P[6] | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1347/2016/G9P[6] | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HGM1883/2018/G9P[6] | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1595/2017/G9P[4] | G9 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E6 | H2 |
| RVA/Human-wt/MOZ/HCN1855/2017/G9P[4] | G9 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1598/2017/G9P[4] | G9 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HGJM1647/2017/G9P[4] | G9 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1600/2017/G9P[4] | G9 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HCN1604/2017/G9P[4] | G9 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/MOZ/HGM483/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGJM0318/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGJM0334/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGM0355/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HCN0370/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGJM0407/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGJM0385/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGM0322/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGJM0497/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGJM0413/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGM0353/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGM0389/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGJM0643/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVA/Human-wt/MOZ/HGJM0644/2015/G9P[8] | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munlela, B.; João, E.D.; Strydom, A.; Bauhofer, A.F.L.; Chissaque, A.; Chilaúle, J.J.; Maurício, I.L.; Donato, C.M.; O’Neill, H.G.; de Deus, N. Whole-Genome Characterization of Rotavirus G9P[6] and G9P[4] Strains That Emerged after Rotavirus Vaccine Introduction in Mozambique. Viruses 2024, 16, 1140. https://doi.org/10.3390/v16071140
Munlela B, João ED, Strydom A, Bauhofer AFL, Chissaque A, Chilaúle JJ, Maurício IL, Donato CM, O’Neill HG, de Deus N. Whole-Genome Characterization of Rotavirus G9P[6] and G9P[4] Strains That Emerged after Rotavirus Vaccine Introduction in Mozambique. Viruses. 2024; 16(7):1140. https://doi.org/10.3390/v16071140
Chicago/Turabian StyleMunlela, Benilde, Eva D. João, Amy Strydom, Adilson Fernando Loforte Bauhofer, Assucênio Chissaque, Jorfélia J. Chilaúle, Isabel L. Maurício, Celeste M. Donato, Hester G. O’Neill, and Nilsa de Deus. 2024. "Whole-Genome Characterization of Rotavirus G9P[6] and G9P[4] Strains That Emerged after Rotavirus Vaccine Introduction in Mozambique" Viruses 16, no. 7: 1140. https://doi.org/10.3390/v16071140
APA StyleMunlela, B., João, E. D., Strydom, A., Bauhofer, A. F. L., Chissaque, A., Chilaúle, J. J., Maurício, I. L., Donato, C. M., O’Neill, H. G., & de Deus, N. (2024). Whole-Genome Characterization of Rotavirus G9P[6] and G9P[4] Strains That Emerged after Rotavirus Vaccine Introduction in Mozambique. Viruses, 16(7), 1140. https://doi.org/10.3390/v16071140