
Detection of Known and Novel Virus Sequences in the Black Solider Fly and Expression of Host Antiviral Pathways


Abstract
:1. Introduction
2. Materials and Methods
2.1. BSF and Substrate Sampling
2.2. RNA Isolation
2.3. Library Preparation and Sequencing
2.4. Metatranscriptome Assembly
2.5. Identification of Virus Sequences
2.6. Phylogenetic Analysis of Novel Virus Sequences
2.7. Overlap of Virus Occurrence across Samples
2.8. Quantification of BSF and Viral Transcripts
2.9. Identifying Candidate Antiviral Genes in BSF
3. Results
3.1. Hermetia illucens Sigma-like Virus 1

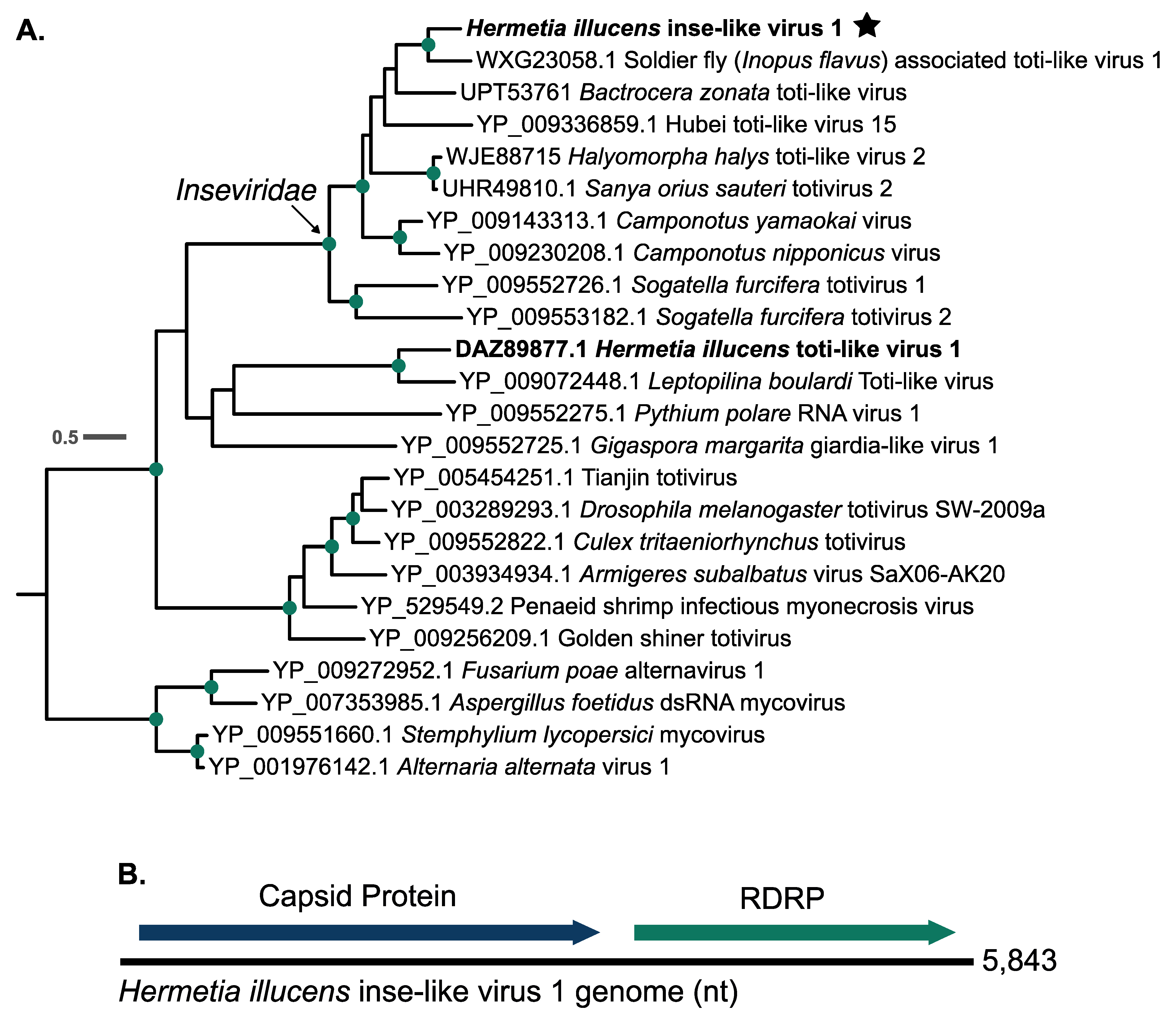
3.2. Hermetia illucens Inse-like Virus 1
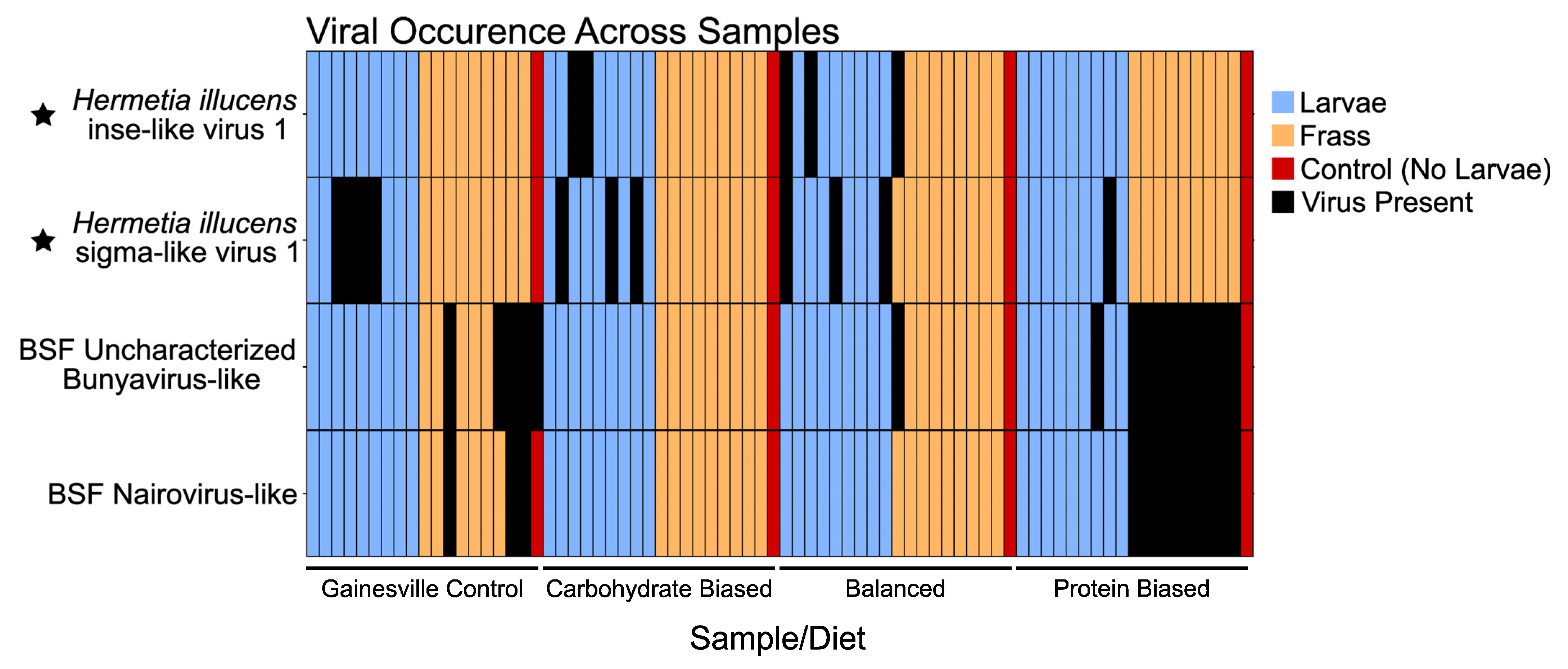
3.3. Known BSF-Associated Viruses
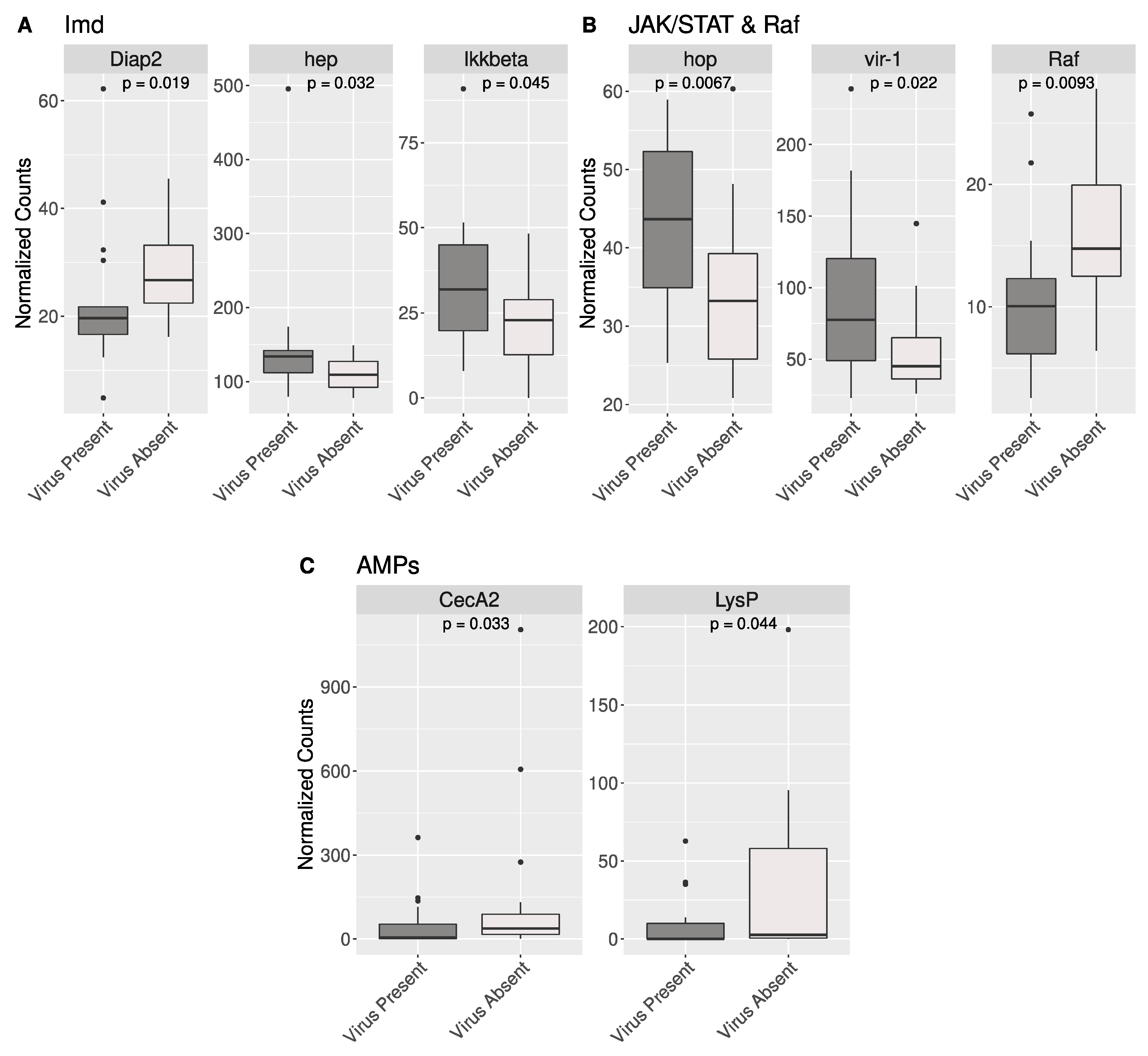
3.4. Antiviral Gene Expression in BSF
4. Discussion
4.1. Hermetia illucens Sigma-like Virus 1
4.2. Hermetia illucens Inse-like Virus 1
4.3. Known BSF Viruses Detected in Our Study
4.4. Gene Expression in Putative Antiviral Genes in BSF
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomberlin, J.K.; van Huis, A. Black Soldier Fly from Pest to “crown Jewel” of the Insects as Feed Industry: An Historical Perspective. J. Insects Food Feed 2020, 6, 1–4. [Google Scholar] [CrossRef]
- van Huis, A. Insects as Food and Feed, a New Emerging Agricultural Sector: A Review. J. Insects Food Feed 2020, 6, 27–44. [Google Scholar] [CrossRef]
- Maciel-Vergara, G.; Ros, V.I.D. Viruses of Insects Reared for Food and Feed. J. Invertebr. Pathol. 2017, 147, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Fang, G.; Cai, M.; Kou, Z.; Xu, J.; Cao, Y.; Bai, L.; Zhang, Y.; Jiang, Y.; Luo, X.; et al. Genomic Landscape and Genetic Manipulation of the Black Soldier Fly Hermetia Illucens, a Natural Waste Recycler. Cell Res. 2020, 30, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Salvia, R.; Scieuzo, C.; Di Somma, A.; Vogel, H.; Pucci, P.; Sgambato, A.; Wolff, M.; Falabella, P. A Bioinformatic Study of Antimicrobial Peptides Identified in the Black Soldier Fly (BSF) Hermetia illucens (Diptera: Stratiomyidae). Sci. Rep. 2020, 10, 16875. [Google Scholar] [CrossRef] [PubMed]
- Vogel, H.; Müller, A.; Heckel, D.G.; Gutzeit, H.; Vilcinskas, A. Nutritional Immunology: Diversification and Diet-Dependent Expression of Antimicrobial Peptides in the Black Soldier Fly Hermetia Illucens. Dev. Comp. Immunol. 2018, 78, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Walt, H.K.; Kooienga, E.; Cammack, J.A.; Tomberlin, J.K.; Jordan, H.R.; Meyer, F.; Hoffmann, F.G. Bioinformatic Surveillance Leads to Discovery of Two Novel Putative Bunyaviruses Associated with Black Soldier Fly. Viruses 2023, 15, 1654. [Google Scholar] [CrossRef] [PubMed]
- Pienaar, R.D.; Gilbert, C.; Belliardo, C.; Herrero, S.; Herniou, E.A. First Evidence of Past and Present Interactions between Viruses and the Black Soldier Fly, Hermetia Illucens. Viruses 2022, 14, 1274. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus Taxonomy: The Database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef]
- Andrews, S. FastQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 28 March 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Generalovic, T.N.; McCarthy, S.A.; Warren, I.A.; Wood, J.M.D.; Torrance, J.; Sims, Y.; Quail, M.; Howe, K.; Pipan, M.; Durbin, R.; et al. A High-Quality, Chromosome-Level Genome Assembly of the Black Soldier Fly (Hermetia illucens L.). G3 Genes Genomes Genet. 2021, 11, jkab085. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Merkel, D. Docker: Lightweight Linux Containers for Consistent Development and Deployment. Linux J. 2014, 2014, 2. [Google Scholar]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein Domains Identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gil, M.; Dufayard, J.F.; Dessimoz, C.; Gascuel, O. Survey of Branch Support Methods Demonstrates Accuracy, Power, and Robustness of Fast Likelihood-Based Approximation Schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention; Environmental Protection Agency. Joint Statement on Bed Bug Control in the United States from the U.S. Centers for Disease Control and Prevention (CDC) and the U.S. Environmental Protection Agency (EPA); U.S. Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2010.
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Rosendo Machado, S.; van der Most, T.; Miesen, P. Genetic Determinants of Antiviral Immunity in Dipteran Insects—Compiling the Experimental Evidence. Dev. Comp. Immunol. 2021, 119, 104010. [Google Scholar] [CrossRef]
- Longdon, B.; Obbard, D.J.; Jiggins, F.M. Sigma Viruses from Three Species of Drosophila Form a Major New Clade in the Rhabdovirus Phylogeny. Proc. Biol. Sci. 2010, 277, 35–44. [Google Scholar] [CrossRef]
- Divekar, G.; Colmant, A.M.G.; Furlong, M.J.; Etebari, K. Transcriptome Analysis Reveals a Diverse Range of Novel Viruses in Australian Sugarcane Soldier Fly (Inopus flavus) Larvae. Viruses 2024, 16, 516. [Google Scholar] [CrossRef]
- Longdon, B.; Wilfert, L.; Jiggins, F.M. The Sigma Viruses of Drosophila. In Rhabdoviruses: Molecular Taxonomy, Evolution, Genomics, Ecology, Host-Vector Interactions, Cytopathology and Control; Dietzgen, R.G., Kuzmin, I.V., Eds.; Caister Academic Press: Poole, UK, 2012; pp. 117–132. [Google Scholar]
- Longdon, B.; Murray, G.G.R.; Palmer, W.J.; Day, J.P.; Parker, D.J.; Welch, J.J.; Obbard, D.J.; Jiggins, F.M. The Evolution, Diversity, and Host Associations of Rhabdoviruses. Virus Evol. 2015, 1, vev014. [Google Scholar] [CrossRef]
- Litov, A.G.; Belova, O.A.; Kholodilov, I.S.; Gadzhikurbanov, M.N.; Gmyl, L.V.; Oorzhak, N.D.; Saryglar, A.A.; Ishmukhametov, A.A.; Karganova, G.G. Possible Arbovirus Found in Virome of Melophagus Ovinus. Viruses 2021, 13, 2375. [Google Scholar] [CrossRef] [PubMed]
- Duxbury, E.M.L.; Day, J.P.; Maria Vespasiani, D.; Thüringer, Y.; Tolosana, I.; Smith, S.C.L.; Tagliaferri, L.; Kamacioglu, A.; Lindsley, I.; Love, L.; et al. Host-Pathogen Coevolution Increases Genetic Variation in Susceptibility to Infection. eLife 2019, 8, e46440. [Google Scholar] [CrossRef]
- Wayne, M.L.; Blohm, G.M.; Brooks, M.E.; Regan, K.L.; Brown, B.Y.; Barfield, M.; Holt, R.D.; Bolker, B.M. The Prevalence and Persistence of Sigma Virus, a Biparentally Transmitted Parasite of Drosophila Melanogaster. Evol. Ecol. Res. 2011, 13, 323–345. [Google Scholar] [PubMed]
- Yampolsky, L.Y.; Webb, C.T.; Shabalina, S.A.; Kondrashov, A.S. Rapid Accumulation of a Vertically Transmitted Parasite Triggered by Relaxation of Natural Selection among Hosts. Evol. Ecol. Res. 1999, 1, 581–589. [Google Scholar]
- Fleuriet, A. Polymorphism of the Drosophila Melanogaster—Sigma Virus System. J. Evol. Biol. 1996, 9, 471–484. [Google Scholar] [CrossRef]
- Fleuriet, A. Comparison of Various Physiological Traits in Flies (Drosophila melanogaster) of Wild Origin, Infected or Uninfected by the Hereditary Rhabdovirus Sigma. Arch. Virol. 1981, 69, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Rittschof, C.C.; Pattanaik, S.; Johnson, L.; Matos, L.F.; Brusini, J.; Wayne, M.L. Sigma Virus and Male Reproductive Success in Drosophila Melanogaster. Behav. Ecol. Sociobiol. 2013, 67, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.-C.; Wang, Z.-G.; Gu, Q.-Y.; Niu, J.-Z.; Wang, J.-J. The Diversity of Viral Community in Invasive Fruit Flies (Bactrocera and Zeugodacus) Revealed by Meta-Transcriptomics. Microb. Ecol. 2022, 83, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the Invertebrate RNA Virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Tighe, A.J.; Ruane, N.M.; Carlsson, J. Potential Origins of Fish Toti-like Viruses in Invertebrates. J. Gen. Virol. 2022, 103, 001775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, W.; Cao, M.; Massart, S.; Wang, X. Two Novel Totiviruses in the White-Backed Planthopper, Sogatella Furcifera. J. Gen. Virol. 2018, 99, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Urayama, S.-I.; Ohmatsu, T.; Sassa, Y.; Sakai, C.; Takata, M.; Hayashi, S.; Nagai, M.; Furuya, T.; Moriyama, H.; et al. Identification, Characterization and Full-Length Sequence Analysis of a Novel DsRNA Virus Isolated from the Arboreal Ant Camponotus Yamaokai. J. Gen. Virol. 2015, 96, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Sakai, C.; Thomas, C.E.; Nunoura, T.; Urayama, S.-I. A New Member of the Family Totiviridae Associated with Arboreal Ants (Camponotus nipponicus). Arch. Virol. 2016, 161, 2043–2045. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Guo, X.; Zhang, S.; Zhao, Q.; Sun, Q.; Zhou, H.; Zhang, J.; Tong, Y. Discovery of Two Novel Totiviruses from Culex Tritaeniorhynchus Classifiable in a Distinct Clade with Arthropod-Infecting Viruses within the Family Totiviridae. Arch. Virol. 2018, 163, 2899–2902. [Google Scholar] [CrossRef]
- Giovannini, L.; Mazza, G.; Chitarra, W.; Sabbatini-Peverieri, G.; Sonnati, C.; Roversi, P.F.; Nerva, L. New Insights from the Virome of Halyomorpha Halys (Stål, 1855). Virus Res. 2022, 316, 198802. [Google Scholar] [CrossRef]
- Martinez, J.; Lepetit, D.; Ravallec, M.; Fleury, F.; Varaldi, J. Additional Heritable Virus in the Parasitic Wasp Leptopilina Boulardi: Prevalence, Transmission and Phenotypic Effects. J. Gen. Virol. 2016, 97, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Jan, E.; Sarnow, P.; Schneider, D. The Imd Pathway Is Involved in Antiviral Immune Responses in Drosophila. PLoS ONE 2009, 4, e7436. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Jouanguy, E.; Irving, P.; Troxler, L.; Galiana-Arnoux, D.; Hetru, C.; Hoffmann, J.A.; Imler, J.-L. The Jak-STAT Signaling Pathway Is Required but Not Sufficient for the Antiviral Response of Drosophila. Nat. Immunol. 2005, 6, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.; Hutter, S.; Baines, J.F.; Roller, J.; Saminadin-Peter, S.S.; Parsch, J.; Jiggins, F.M. The Transcriptional Response of Drosophila Melanogaster to Infection with the Sigma Virus (Rhabdoviridae). PLoS ONE 2009, 4, e6838. [Google Scholar] [CrossRef]
- Ballinger, M.J.; Christian, R.C.; Moore, L.D.; Taylor, D.J.; Sabet, A. Evolution and Diversity of Inherited Viruses in the Nearctic Phantom Midge, Chaoborus Americanus. Virus Evol. 2022, 8, veac018. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BSF Gene | Drosophila Ortholog | Function in Drosophila |
|---|---|---|
| LOC119654977 | Diap2 | Mediator of NF-kB signaling—required for innate immune response |
| LOC119653405 | hep | Critical for JNK activation in immune signaling |
| LOC119646668 | IKKbeta | Regulates antiviral response in Imd |
| LOC119651484 | hop | Induces expression of JAK/STAT regulated genes |
| LOC119646570 | vir-1 | Regulated by JAK/STAT; expressed in response to viral infection |
| LOC119656159 | Raf | Component of Ras/Raf pathway; interacts with hop of JAK/STAT |
| LOC119653270 | CecA2 | Antimicrobial peptide |
| LOC119654763 | LysP | Antimicrobial activity against Gram-negative bacteria |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walt, H.K.; Jordan, H.R.; Meyer, F.; Hoffmann, F.G. Detection of Known and Novel Virus Sequences in the Black Solider Fly and Expression of Host Antiviral Pathways. Viruses 2024, 16, 1219. https://doi.org/10.3390/v16081219
Walt HK, Jordan HR, Meyer F, Hoffmann FG. Detection of Known and Novel Virus Sequences in the Black Solider Fly and Expression of Host Antiviral Pathways. Viruses. 2024; 16(8):1219. https://doi.org/10.3390/v16081219
Chicago/Turabian StyleWalt, Hunter K., Heather R. Jordan, Florencia Meyer, and Federico G. Hoffmann. 2024. "Detection of Known and Novel Virus Sequences in the Black Solider Fly and Expression of Host Antiviral Pathways" Viruses 16, no. 8: 1219. https://doi.org/10.3390/v16081219
APA StyleWalt, H. K., Jordan, H. R., Meyer, F., & Hoffmann, F. G. (2024). Detection of Known and Novel Virus Sequences in the Black Solider Fly and Expression of Host Antiviral Pathways. Viruses, 16(8), 1219. https://doi.org/10.3390/v16081219