
No Time to Die: How Cytomegaloviruses Suppress Apoptosis, Necroptosis, and Pyroptosis



Abstract
1. Cytomegalovirus Infection and Pathogenesis
2. Overview of Programmed Cell Death Pathways Activated during CMV Infection
2.1. Inhibition of Apoptosis by CMVs
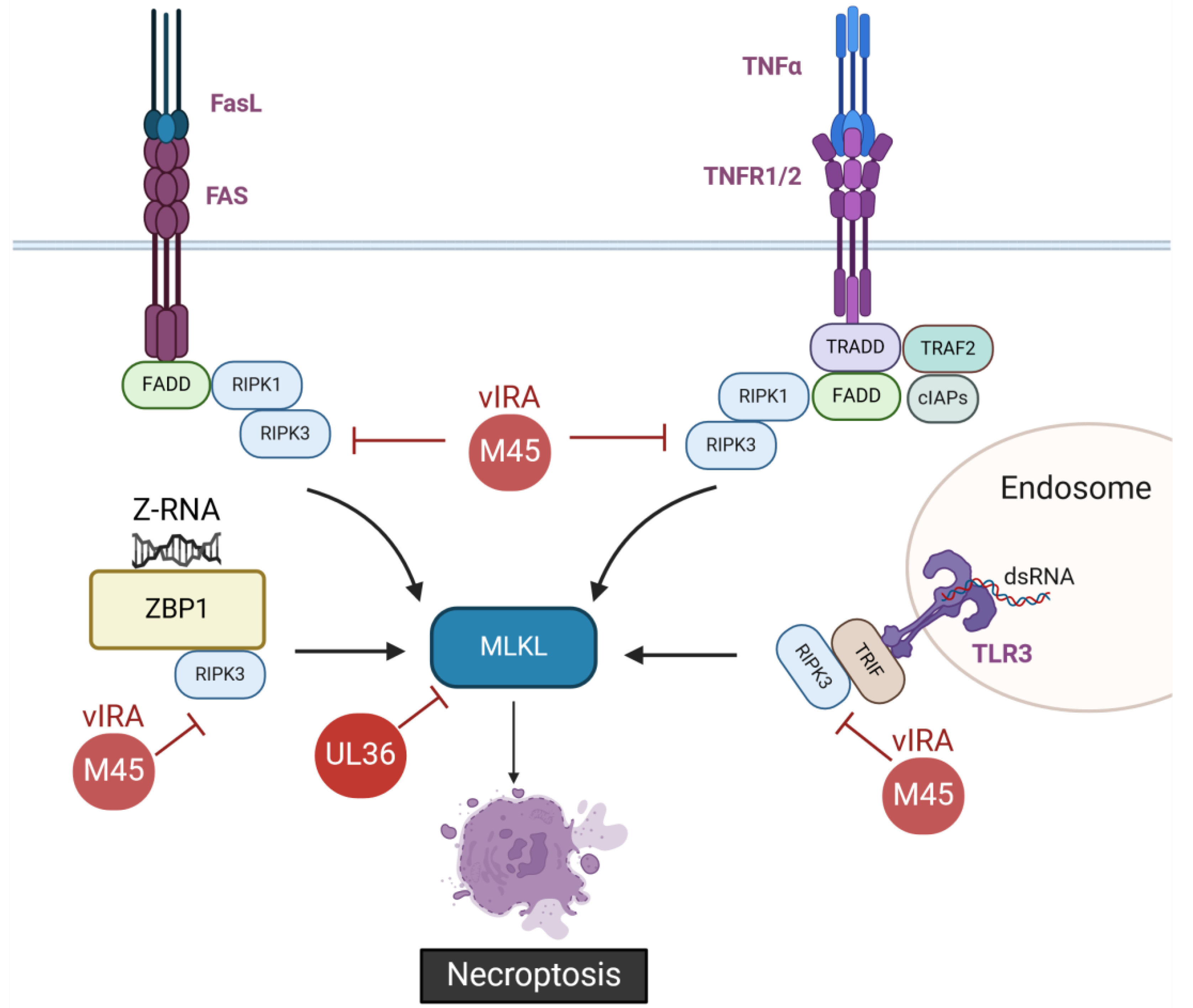
2.2. Inhibition of Necroptosis by CMVs
2.3. Inhibition of Pyroptosis by CMVs
3. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Mocarski, E.S., Jr. Immunomodulation by cytomegaloviruses: Manipulative strategies beyond evasion. Trends Microbiol. 2002, 10, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Hengel, H.; Brune, W.; Koszinowski, U.H. Immune evasion by cytomegalovirus--survival strategies of a highly adapted opportunist. Trends Microbiol. 1998, 6, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Patro, A.R.K. Subversion of Immune Response by Human Cytomegalovirus. Front. Immunol. 2019, 10, 1155. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Brune, W. Molecular Basis of Cytomegalovirus Host Species Specificity. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M., Ed.; Caister Academic Press: Wymondham, UK, 2013; Volume 1, pp. 322–329. [Google Scholar]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [PubMed]
- Gerna, G.; Kabanova, A.; Lilleri, D. Human Cytomegalovirus Cell Tropism and Host Cell Receptors. Vaccines 2019, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.; Watson, G.M.; Jonjic, S.; Degli-Esposti, M.A.; Rossjohn, J. Modulation of innate and adaptive immunity by cytomegaloviruses. Nat. Rev. Immunol. 2020, 20, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Brune, W.; Andoniou, C.E. Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses 2017, 9, 249. [Google Scholar] [CrossRef]
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Apoptosis, Pyroptosis, and Necroptosis-Oh My! The Many Ways a Cell Can Die. J. Mol. Biol. 2022, 434, 167378. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- de Vasconcelos, N.M.; Lamkanfi, M. Recent Insights on Inflammasomes, Gasdermin Pores, and Pyroptosis. Cold Spring Harb. Perspect. Biol. 2020, 12, a036392. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Chan, F.K. Staying alive: Cell death in antiviral immunity. Mol. Cell 2014, 54, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Kang, T.B.; Dillon, C.P.; Green, D.R. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 2016, 352, aaf2154. [Google Scholar] [CrossRef] [PubMed]
- Place, D.E.; Lee, S.; Kanneganti, T.D. PANoptosis in microbial infection. Curr. Opin. Microbiol. 2021, 59, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Lee, S. Recent advances in ZBP1-derived PANoptosis against viral infections. Front. Immunol. 2023, 14, 1148727. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M. Signalling by proteolysis: Death receptors induce apoptosis. Int. J. Clin. Lab. Res. 1998, 28, 141–147. [Google Scholar] [CrossRef]
- Wajant, H. Death receptors. Essays Biochem. 2003, 39, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Choi, E.J.; Joe, C.O. Activation of death-inducing signaling complex (DISC) by pro-apoptotic C-terminal fragment of RIP. Oncogene 2000, 19, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chun, H.J.; Wong, W.; Spencer, D.M.; Lenardo, M.J. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 13884–13888. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.H.; Goeddel, D.V. Fas antigen and p55 TNF receptor signal apoptosis through distinct pathways. J. Immunol. 1994, 152, 1751–1755. [Google Scholar] [CrossRef]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Boldin, M.P.; Varfolomeev, E.E.; Pancer, Z.; Mett, I.L.; Camonis, J.H.; Wallach, D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 1995, 270, 7795–7798. [Google Scholar] [CrossRef]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Glover, H.L.; Schreiner, A.; Dewson, G.; Tait, S.W.G. Mitochondria and cell death. Nat. Cell Biol. 2024. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes. Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Meusburger, S.; Hawkins, C.J.; Riglar, D.T.; Lee, E.F.; Fairlie, W.D.; Huang, D.C.; Adams, J.M. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl. Acad. Sci. USA 2008, 105, 18081–18087. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Westphal, D.; Dewson, G.; Ma, S.; Hockings, C.; Fairlie, W.D.; Lee, E.F.; Yao, S.; Robin, A.Y.; Smith, B.J.; et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 2013, 152, 519–531. [Google Scholar] [CrossRef]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Datta, P.; Fan, X.J.; Fernandes-Alnemri, T.; Huang, Z.; Alnemri, E.S. Molecular determinants of the caspase-promoting activity of Smac/DIABLO and its role in the death receptor pathway. J. Biol. Chem. 2000, 275, 36152–36157. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef]
- Bao, Q.; Shi, Y. Apoptosome: A platform for the activation of initiator caspases. Cell Death Differ. 2007, 14, 56–65. [Google Scholar] [CrossRef]
- Johnston, B.P.; McCormick, C. Herpesviruses and the Unfolded Protein Response. Viruses 2019, 12, 17. [Google Scholar] [CrossRef]
- Terhune, S.; Torigoi, E.; Moorman, N.; Silva, M.; Qian, Z.; Shenk, T.; Yu, D. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 2007, 81, 3109–3123. [Google Scholar] [CrossRef]
- Xuan, B.; Qian, Z.; Torigoi, E.; Yu, D. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 2009, 83, 3463–3474. [Google Scholar] [CrossRef]
- Qian, Z.; Xuan, B.; Gualberto, N.; Yu, D. The human cytomegalovirus protein pUL38 suppresses endoplasmic reticulum stress-mediated cell death independently of its ability to induce mTORC1 activation. J. Virol. 2011, 85, 9103–9113. [Google Scholar] [CrossRef]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef]
- Verma, S.; Loewendorf, A.; Wang, Q.; McDonald, B.; Redwood, A.; Benedict, C.A. Inhibition of the TRAIL death receptor by CMV reveals its importance in NK cell-mediated antiviral defense. PLoS Pathog. 2014, 10, e1004268. [Google Scholar] [CrossRef]
- Smith, W.; Tomasec, P.; Aicheler, R.; Loewendorf, A.; Nemcovicova, I.; Wang, E.C.; Stanton, R.J.; Macauley, M.; Norris, P.; Willen, L.; et al. Human cytomegalovirus glycoprotein UL141 targets the TRAIL death receptors to thwart host innate antiviral defenses. Cell Host Microbe 2013, 13, 324–335. [Google Scholar] [CrossRef]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar] [CrossRef]
- McCormick, A.L.; Skaletskaya, A.; Barry, P.A.; Mocarski, E.S.; Goldmacher, V.S. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 2003, 316, 221–233. [Google Scholar] [CrossRef]
- Menard, C.; Wagner, M.; Ruzsics, Z.; Holak, K.; Brune, W.; Campbell, A.E.; Koszinowski, U.H. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J. Virol. 2003, 77, 5557–5570. [Google Scholar] [CrossRef]
- Chaudhry, M.Z.; Kasmapour, B.; Plaza-Sirvent, C.; Bajagic, M.; Casalegno Garduno, R.; Borkner, L.; Lenac Rovis, T.; Scrima, A.; Jonjic, S.; Schmitz, I.; et al. UL36 Rescues Apoptosis Inhibition and In vivo Replication of a Chimeric MCMV Lacking the M36 Gene. Front. Cell Infect. Microbiol. 2017, 7, 312. [Google Scholar] [CrossRef]
- Cicin-Sain, L.; Ruzsics, Z.; Podlech, J.; Bubic, I.; Menard, C.; Jonjic, S.; Reddehase, M.J.; Koszinowski, U.H. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J. Virol. 2008, 82, 2056–2064. [Google Scholar] [CrossRef]
- Chiou, S.H.; Liu, J.H.; Hsu, W.M.; Chen, S.S.; Chang, S.Y.; Juan, L.J.; Lin, J.C.; Yang, Y.T.; Wong, W.W.; Liu, C.Y.; et al. Up-regulation of Fas ligand expression by human cytomegalovirus immediate-early gene product 2: A novel mechanism in cytomegalovirus-induced apoptosis in human retina. J. Immunol. 2001, 167, 4098–4103. [Google Scholar] [CrossRef]
- Chiou, S.H.; Yang, Y.P.; Lin, J.C.; Hsu, C.H.; Jhang, H.C.; Yang, Y.T.; Lee, C.H.; Ho, L.L.; Hsu, W.M.; Ku, H.H.; et al. The immediate early 2 protein of human cytomegalovirus (HCMV) mediates the apoptotic control in HCMV retinitis through up-regulation of the cellular FLICE-inhibitory protein expression. J. Immunol. 2006, 177, 6199–6206. [Google Scholar] [CrossRef]
- Goldmacher, V.S.; Bartle, L.M.; Skaletskaya, A.; Dionne, C.A.; Kedersha, N.L.; Vater, C.A.; Han, J.W.; Lutz, R.J.; Watanabe, S.; Cahir McFarland, E.D.; et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA 1999, 96, 12536–12541. [Google Scholar] [CrossRef]
- Arnoult, D.; Bartle, L.M.; Skaletskaya, A.; Poncet, D.; Zamzami, N.; Park, P.U.; Sharpe, J.; Youle, R.J.; Goldmacher, V.S. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. USA 2004, 101, 7988–7993. [Google Scholar] [CrossRef]
- Poncet, D.; Larochette, N.; Pauleau, A.L.; Boya, P.; Jalil, A.A.; Cartron, P.F.; Vallette, F.; Schnebelen, C.; Bartle, L.M.; Skaletskaya, A.; et al. An anti-apoptotic viral protein that recruits Bax to mitochondria. J. Biol. Chem. 2004, 279, 22605–22614. [Google Scholar] [CrossRef]
- Norris, K.L.; Youle, R.J. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J. Virol. 2008, 82, 6232–6243. [Google Scholar] [CrossRef]
- McCormick, A.L.; Roback, L.; Mocarski, E.S. HtrA2/Omi terminates cytomegalovirus infection and is controlled by the viral mitochondrial inhibitor of apoptosis (vMIA). PLoS Pathog. 2008, 4, e1000063. [Google Scholar] [CrossRef]
- Magalhaes, A.C.; Ferreira, A.R.; Gomes, S.; Vieira, M.; Gouveia, A.; Valenca, I.; Islinger, M.; Nascimento, R.; Schrader, M.; Kagan, J.C.; et al. Peroxisomes are platforms for cytomegalovirus’ evasion from the cellular immune response. Sci. Rep. 2016, 6, 26028. [Google Scholar] [CrossRef]
- Jurak, I.; Schumacher, U.; Simic, H.; Voigt, S.; Brune, W. Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J. Virol. 2008, 82, 4812–4822. [Google Scholar] [CrossRef]
- Arnoult, D.; Skaletskaya, A.; Estaquier, J.; Dufour, C.; Goldmacher, V.S. The murine cytomegalovirus cell death suppressor m38.5 binds Bax and blocks Bax-mediated mitochondrial outer membrane permeabilization. Apoptosis 2008, 13, 1100–1110. [Google Scholar] [CrossRef]
- Manzur, M.; Fleming, P.; Huang, D.C.; Degli-Esposti, M.A.; Andoniou, C.E. Virally mediated inhibition of Bax in leukocytes promotes dissemination of murine cytomegalovirus. Cell Death Differ. 2009, 16, 312–320. [Google Scholar] [CrossRef]
- Cam, M.; Handke, W.; Picard-Maureau, M.; Brune, W. Cytomegaloviruses inhibit Bak- and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ. 2010, 17, 655–665. [Google Scholar] [CrossRef]
- Noguchi, K.; Majima, R.; Takahashi, K.; Iwase, Y.; Yamada, S.; Satoh, K.; Koshizuka, T.; Inoue, N. Identification and functional analyses of a cell-death inhibitor encoded by guinea pig cytomegalovirus gp38.1 in cell culture and in animals. J. Gen. Virol. 2020, 101, 1270–1279. [Google Scholar] [CrossRef]
- Satoh, K.; Takahashi, K.; Noguchi, K.; Kobayashi, Y.; Majima, R.; Iwase, Y.; Yamaguchi, K.; Masuda, Y.; Koshizuka, T.; Inoue, N. Characterization of the Second Apoptosis Inhibitor Encoded by Guinea Pig Cytomegalovirus. J. Virol. 2022, 96, e0162222. [Google Scholar] [CrossRef]
- Fleming, P.; Kvansakul, M.; Voigt, V.; Kile, B.T.; Kluck, R.M.; Huang, D.C.; Degli-Esposti, M.A.; Andoniou, C.E. MCMV-mediated inhibition of the pro-apoptotic Bak protein is required for optimal in vivo replication. PLoS Pathog. 2013, 9, e1003192. [Google Scholar] [CrossRef] [PubMed]
- Crosby, L.N.; McCormick, A.L.; Mocarski, E.S. Gene products of the embedded m41/m41.1 locus of murine cytomegalovirus differentially influence replication and pathogenesis. Virology 2013, 436, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Handke, W.; Luig, C.; Popovic, B.; Krmpotic, A.; Jonjic, S.; Brune, W. Viral inhibition of BAK promotes murine cytomegalovirus dissemination to salivary glands. J. Virol. 2013, 87, 3592–3596. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Nagrani, L.N.; Hernandez, L.; McCormick, A.L.; Dillon, C.P.; Koehler, H.S.; Roback, L.; Alnemri, E.S.; Green, D.R.; Mocarski, E.S. Multiple Autonomous Cell Death Suppression Strategies Ensure Cytomegalovirus Fitness. Viruses 2021, 13, 1707. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Strasser, A.; Kayagaki, N.; Dixit, V.M. Cell death. Cell 2024, 187, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.L.; Garnish, S.E.; Hildebrand, J.M.; Murphy, J.M. Location, location, location: A compartmentalized view of TNF-induced necroptotic signaling. Sci. Signal 2021, 14, eabc6178. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J. DAI Another Way: Necroptotic Control of Viral Infection. Cell Host Microbe 2017, 21, 290–293. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 2008, 283, 16966–16970. [Google Scholar] [CrossRef]
- Lembo, D.; Brune, W. Tinkering with a viral ribonucleotide reductase. Trends Biochem. Sci. 2009, 34, 25–32. [Google Scholar] [CrossRef]
- Muscolino, E.; Schmitz, R.; Loroch, S.; Caragliano, E.; Schneider, C.; Rizzato, M.; Kim, Y.H.; Krause, E.; Juranic Lisnic, V.; Sickmann, A.; et al. Herpesviruses induce aggregation and selective autophagy of host signalling proteins NEMO and RIPK1 as an immune-evasion mechanism. Nat. Microbiol. 2020, 5, 331–342. [Google Scholar] [CrossRef]
- Muscolino, E.; Luoto, L.M.; Brune, W. Viral Induced Protein Aggregation: A Mechanism of Immune Evasion. Int. J. Mol. Sci. 2021, 22, 9624. [Google Scholar] [CrossRef]
- Mack, C.; Sickmann, A.; Lembo, D.; Brune, W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc. Natl. Acad. Sci. USA 2008, 105, 3094–3099. [Google Scholar] [CrossRef]
- Krause, E.; de Graaf, M.; Fliss, P.M.; Dölken, L.; Brune, W. Murine cytomegalovirus virion-associated protein M45 mediates rapid NF-kappaB activation after infection. J. Virol. 2014, 88, 9963–9975. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef]
- Maelfait, J.; Liverpool, L.; Bridgeman, A.; Ragan, K.B.; Upton, J.W.; Rehwinkel, J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017, 36, 2529–2543. [Google Scholar] [CrossRef]
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral mediated redirection of NEMO/IKKgamma to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar] [CrossRef]
- Lembo, D.; Donalisio, M.; Hofer, A.; Cornaglia, M.; Brune, W.; Koszinowski, U.; Thelander, L.; Landolfo, S. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J. Virol. 2004, 78, 4278–4288. [Google Scholar] [CrossRef]
- Brune, W.; Menard, C.; Heesemann, J.; Koszinowski, U.H. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 2001, 291, 303–305. [Google Scholar] [CrossRef]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, S.Q.; Liang, Y.; Zhou, X.; Chen, W.; Li, L.; Wu, J.; Zhuang, Q.; Chen, C.; Li, J.; et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 2015, 17, 229–242. [Google Scholar] [CrossRef]
- Kwon, K.M.; Oh, S.E.; Kim, Y.E.; Han, T.H.; Ahn, J.H. Cooperative inhibition of RIP1-mediated NF-kappaB signaling by cytomegalovirus-encoded deubiquitinase and inactive homolog of cellular ribonucleotide reductase large subunit. PLoS Pathog. 2017, 13, e1006423. [Google Scholar] [CrossRef]
- Omoto, S.; Guo, H.; Talekar, G.R.; Roback, L.; Kaiser, W.J.; Mocarski, E.S. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J. Biol. Chem. 2015, 290, 11635–11648. [Google Scholar] [CrossRef]
- Fletcher-Etherington, A.; Nobre, L.; Nightingale, K.; Antrobus, R.; Nichols, J.; Davison, A.J.; Stanton, R.J.; Weekes, M.P. Human cytomegalovirus protein pUL36: A dual cell death pathway inhibitor. Proc. Natl. Acad. Sci. USA 2020, 117, 18771–18779. [Google Scholar] [CrossRef]
- Muscolino, E.; Castiglioni, C.; Brixel, R.; Frascaroli, G.; Brune, W. Species-Specific Inhibition of Necroptosis by HCMV UL36. Viruses 2021, 13, 2134. [Google Scholar] [CrossRef]
- Huerfano, S.; Sroller, V.; Brustikova, K.; Hornikova, L.; Forstova, J. The Interplay between Viruses and Host DNA Sensors. Viruses 2022, 14, 666. [Google Scholar] [CrossRef]
- Kumar, A.; Stavrakis, G.; Karaba, A.H. Herpesviruses and Inflammasomes: One Sensor Does Not Fit All. mBio 2022, 13, e0173721. [Google Scholar] [CrossRef]
- Yu, G.; Choi, Y.K.; Lee, S. Inflammasome diversity: Exploring novel frontiers in the innate immune response. Trends Immunol. 2024, 45, 248–258. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef]
- Sborgi, L.; Ravotti, F.; Dandey, V.P.; Dick, M.S.; Mazur, A.; Reckel, S.; Chami, M.; Scherer, S.; Huber, M.; Bockmann, A.; et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 13237–13242. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Degen, M.; Santos, J.C.; Pluhackova, K.; Cebrero, G.; Ramos, S.; Jankevicius, G.; Hartenian, E.; Guillerm, U.; Mari, S.A.; Kohl, B.; et al. Structural basis of NINJ1-mediated plasma membrane rupture in cell death. Nature 2023, 618, 1065–1071. [Google Scholar] [CrossRef]
- David, L.; Borges, J.P.; Hollingsworth, L.R.; Volchuk, A.; Jansen, I.; Garlick, E.; Steinberg, B.E.; Wu, H. NINJ1 mediates plasma membrane rupture by cutting and releasing membrane disks. Cell 2024, 187, 2224–2235.e16. [Google Scholar] [CrossRef]
- Netea, M.G.; Simon, A.; van de Veerdonk, F.; Kullberg, B.J.; Van der Meer, J.W.; Joosten, L.A. IL-1beta processing in host defense: Beyond the inflammasomes. PLoS Pathog. 2010, 6, e1000661. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Sarkar, A.; Vande Walle, L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, L.; Ma, D.; Liao, Y.; Lu, Y.; Huang, H.; Qin, W.; Liu, X.; Fang, F. Human cytomegalovirus triggers the assembly of AIM2 inflammasome in THP-1-derived macrophages. J. Med. Virol. 2017, 89, 2188–2195. [Google Scholar] [CrossRef]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Soby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef]
- Deng, Y.; Ostermann, E.; Brune, W. A cytomegalovirus inflammasome inhibitor reduces proinflammatory cytokine release and pyroptosis. Nat. Commun. 2024, 15, 786. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Brickey, W.J.; Ting, J.P. Inflammasomes in Myeloid Cells: Warriors Within. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Carter, J.J.; Nemeno, J.G.E.; Oh, J.J.; Houghton, J.E.; Dix, R.D. Atypical cytomegalovirus retinal disease in pyroptosis-deficient mice with murine acquired immunodeficiency syndrome. Exp. Eye Res. 2021, 209, 108651. [Google Scholar] [CrossRef] [PubMed]
- Morello, C.S.; Cranmer, L.D.; Spector, D.H. In vivo replication, latency, and immunogenicity of murine cytomegalovirus mutants with deletions in the M83 and M84 genes, the putative homologs of human cytomegalovirus pp65 (UL83). J. Virol. 1999, 73, 7678–7693. [Google Scholar] [CrossRef]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, D.; Huang, H.; Lu, Y.; Liao, Y.; Liu, L.; Liu, X.; Fang, F. Interaction between HCMV pUL83 and human AIM2 disrupts the activation of the AIM2 inflammasome. Virol. J. 2017, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Botto, S.; Abraham, J.; Mizuno, N.; Pryke, K.; Gall, B.; Landais, I.; Streblow, D.N.; Fruh, K.J.; DeFilippis, V.R. Human Cytomegalovirus Immediate Early 86-kDa Protein Blocks Transcription and Induces Degradation of the Immature Interleukin-1beta Protein during Virion-Mediated Activation of the AIM2 Inflammasome. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Brizic, I.; Lisnic, B.; Krstanovic, F.; Brune, W.; Hengel, H.; Jonjic, S. Mouse Models for Cytomegalovirus Infections in Newborns and Adults. Curr. Protoc. 2022, 2, e537. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, M.Z.; Casalegno-Garduno, R.; Sitnik, K.M.; Kasmapour, B.; Pulm, A.K.; Brizic, I.; Eiz-Vesper, B.; Moosmann, A.; Jonjic, S.; Mocarski, E.S.; et al. Cytomegalovirus inhibition of extrinsic apoptosis determines fitness and resistance to cytotoxic CD8 T cells. Proc. Natl. Acad. Sci. USA 2020, 117, 12961–12968. [Google Scholar] [CrossRef] [PubMed]
- Ebermann, L.; Ruzsics, Z.; Guzman, C.A.; van Rooijen, N.; Casalegno-Garduno, R.; Koszinowski, U.; Cicin-Sain, L. Block of death-receptor apoptosis protects mouse cytomegalovirus from macrophages and is a determinant of virulence in immunodeficient hosts. PLoS Pathog. 2012, 8, e1003062. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687.e613. [Google Scholar] [CrossRef]
- Lee, S.; Karki, R.; Wang, Y.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 2021, 597, 415–419. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Apoptosis | Necroptosis | Pyroptosis | |
|---|---|---|---|
| Lytic cell death | No | Yes | Yes |
| Pore-forming protein | BAX/BAK | MLKL | GSDM proteins |
| Caspase activation | Yes | No | Yes |
| Inflammatory response | No | Yes | Yes |
| Morphological features | Cell shrinkage Nuclear condensation Membrane blebbing | Cell swelling Nuclear condensation Membrane rupture | Cell swelling Membrane rupture |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Águeda-Pinto, A.; Brune, W. No Time to Die: How Cytomegaloviruses Suppress Apoptosis, Necroptosis, and Pyroptosis. Viruses 2024, 16, 1272. https://doi.org/10.3390/v16081272
Deng Y, Águeda-Pinto A, Brune W. No Time to Die: How Cytomegaloviruses Suppress Apoptosis, Necroptosis, and Pyroptosis. Viruses. 2024; 16(8):1272. https://doi.org/10.3390/v16081272
Chicago/Turabian StyleDeng, Yingqi, Ana Águeda-Pinto, and Wolfram Brune. 2024. "No Time to Die: How Cytomegaloviruses Suppress Apoptosis, Necroptosis, and Pyroptosis" Viruses 16, no. 8: 1272. https://doi.org/10.3390/v16081272
APA StyleDeng, Y., Águeda-Pinto, A., & Brune, W. (2024). No Time to Die: How Cytomegaloviruses Suppress Apoptosis, Necroptosis, and Pyroptosis. Viruses, 16(8), 1272. https://doi.org/10.3390/v16081272