
Genetic Diversity and Spatiotemporal Distribution of SARS-CoV-2 Variants in Guinea: A Meta-Analysis of Sequence Data (2020–2023)

 , ,
, ,  ,
,  , , , , , , ,
, , , , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Phylogeography Reconstruction
2.3. Analysis of Introduction of VOI and VOC Variants in Guinea
2.4. Mutations Diversity Analysis
3. Results
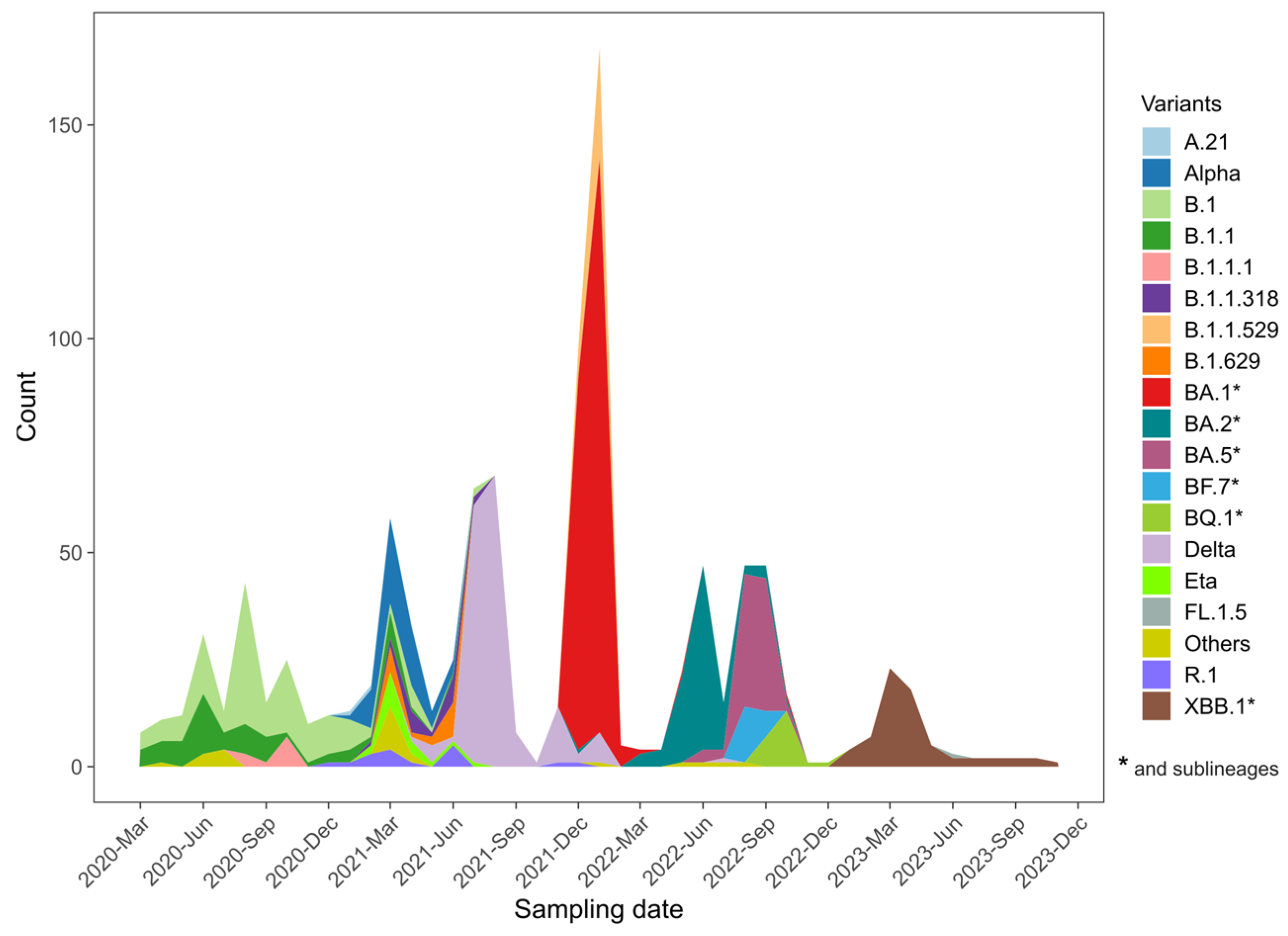
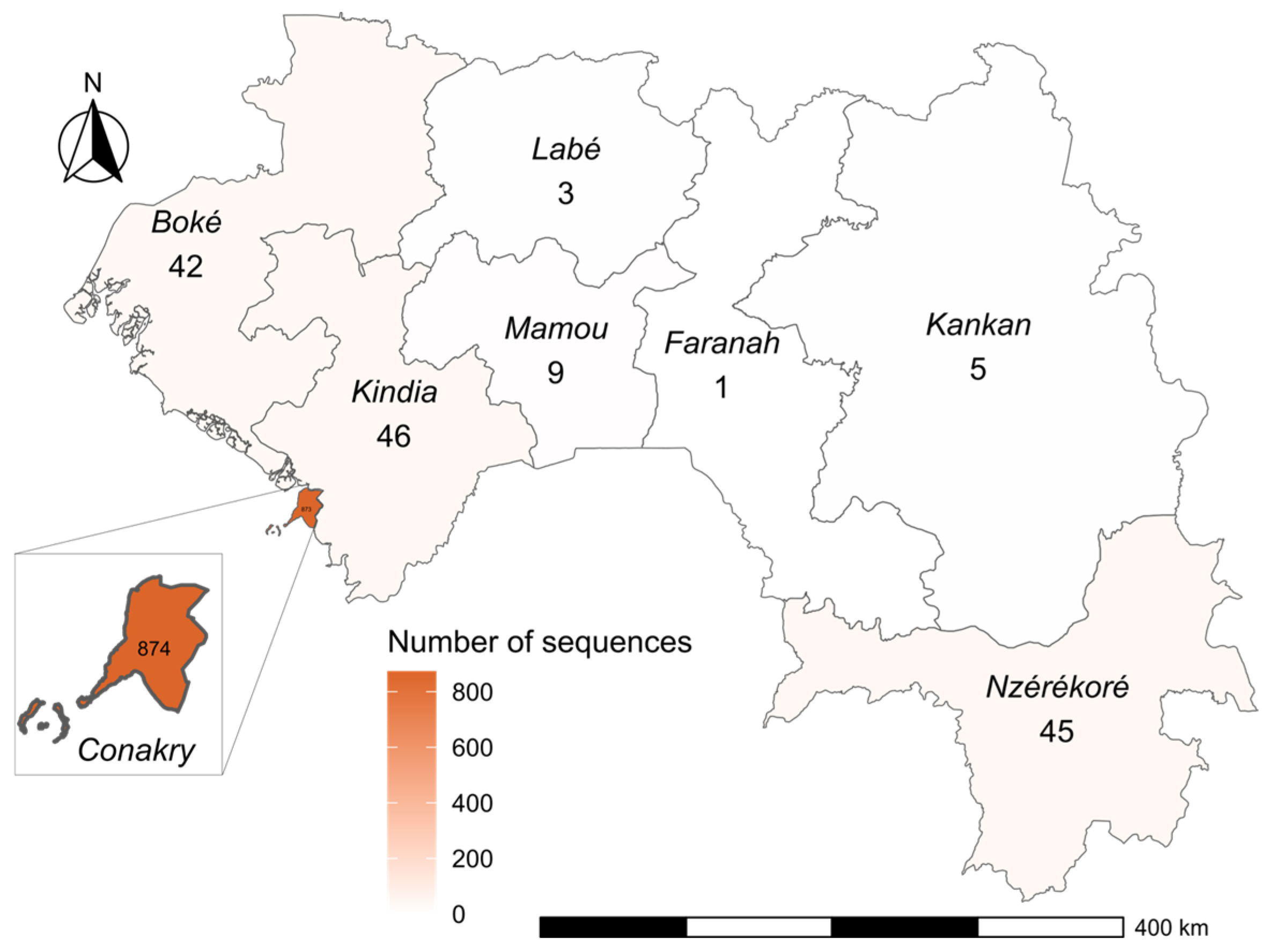
3.1. SARS-CoV-2 Variant Distribution
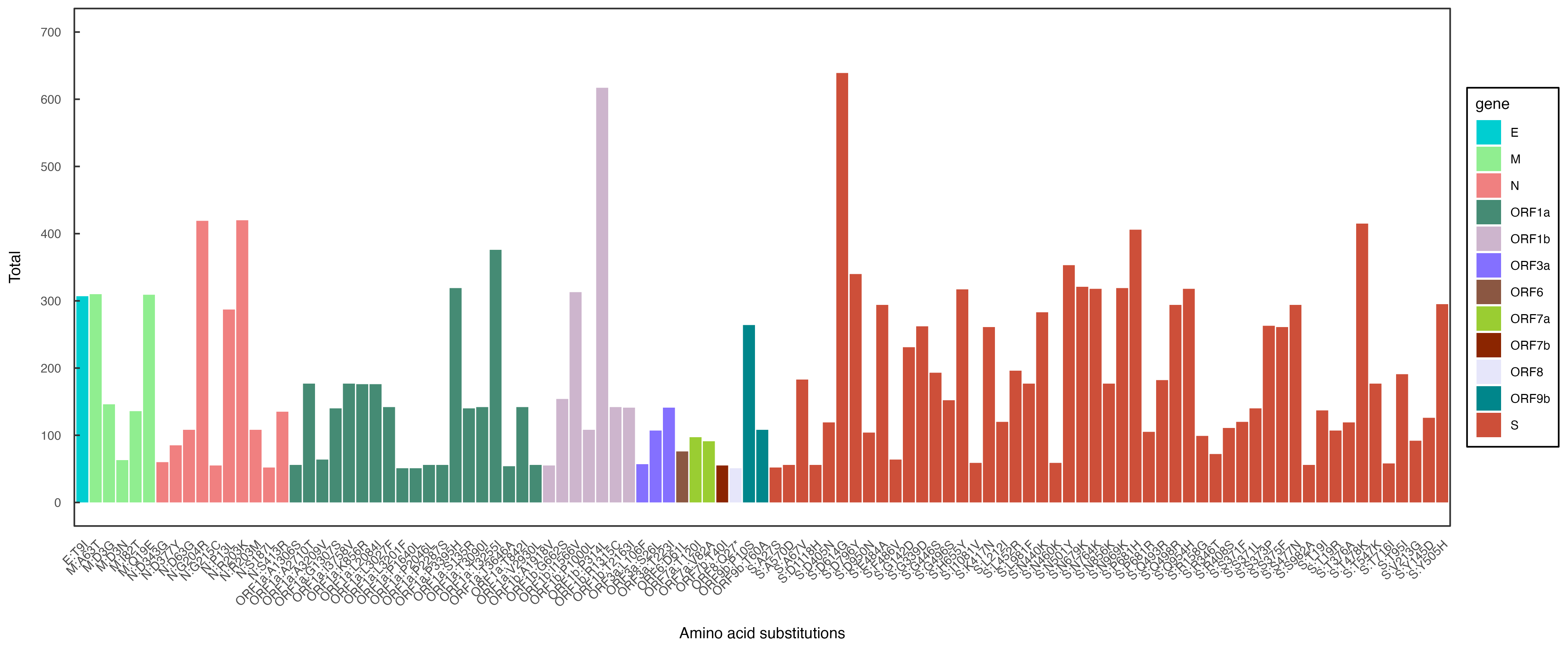
3.2. Mutational Analysis
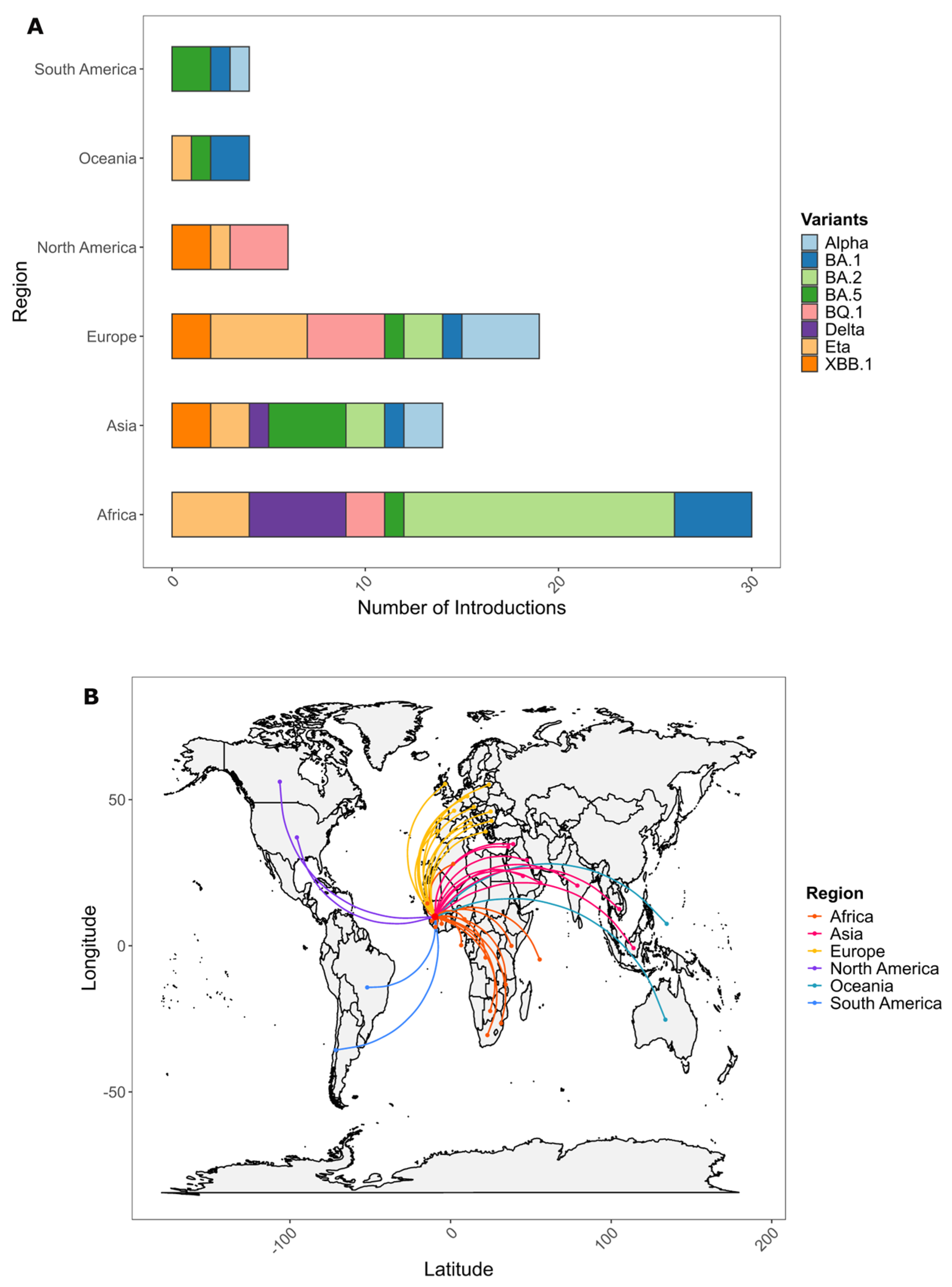
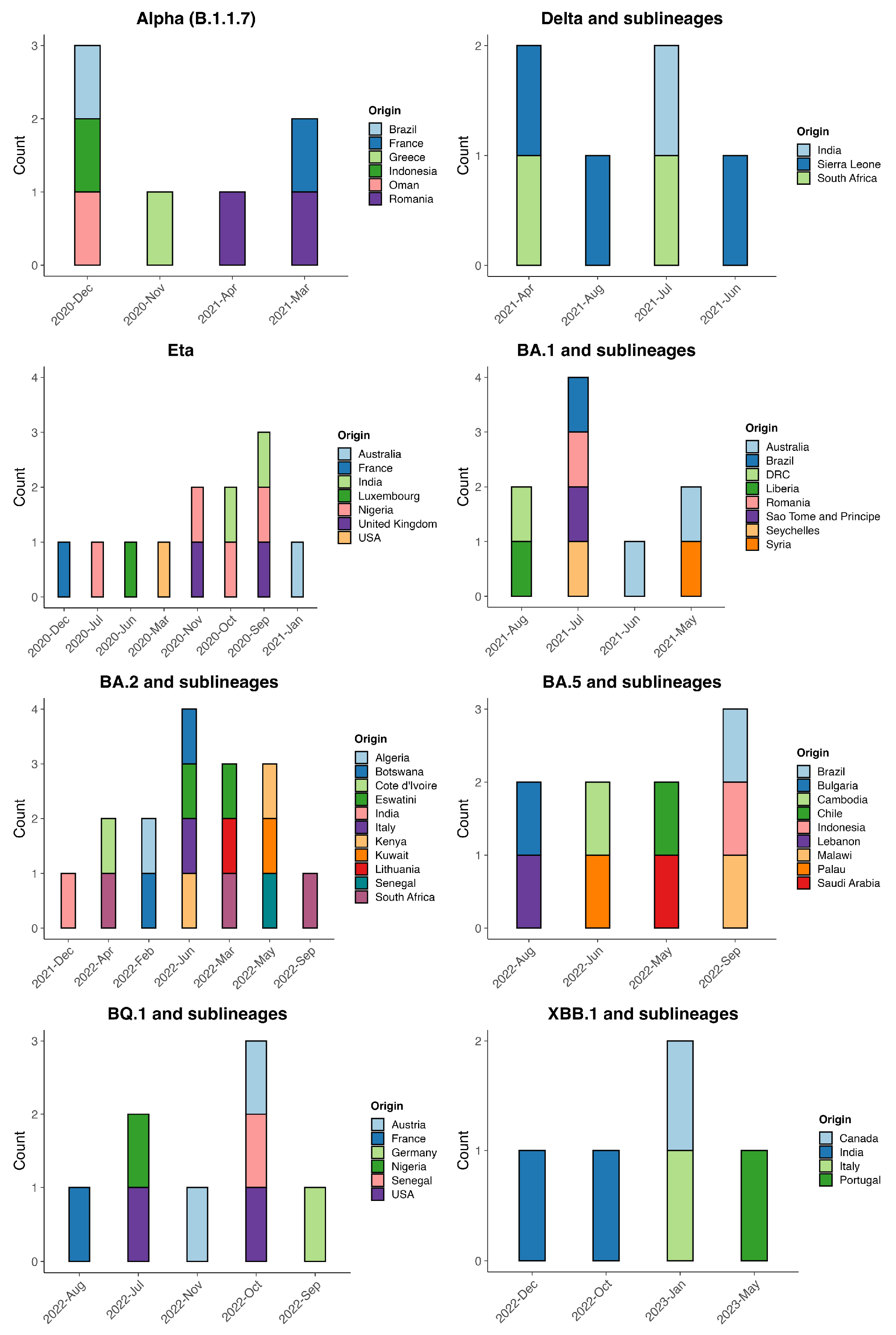
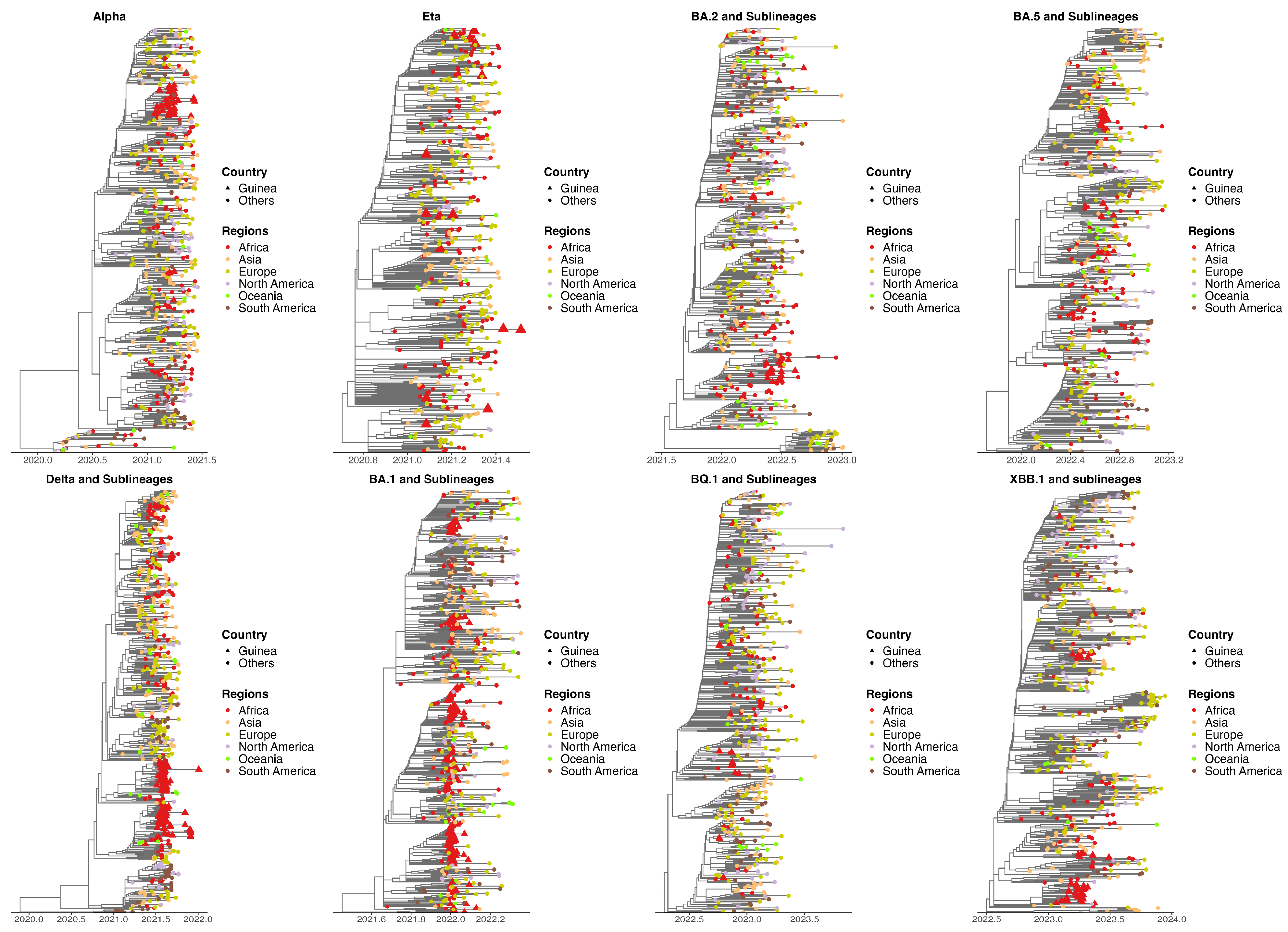
3.3. Origins of Variants of Concern Circulating in Guinea
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flores-Vega, V.R.; Monroy-Molina, J.V.; Jiménez-Hernández, L.E.; Torres, A.G.; Santos-Preciado, J.I.; Rosales-Reyes, R. SARS-CoV-2: Evolution and Emergence of New Viral Variants. Viruses 2022, 14, 653. [Google Scholar] [CrossRef]
- Si, Y.; Wu, W.; Xue, X.; Sun, X.; Qin, Y.; Li, Y.; Qiu, C.; Li, Y.; Zhuo, Z.; Mi, Y.; et al. The evolution of SARS-CoV-2 and the COVID-19 pandemic. PeerJ 2023, 11, e15990. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Fournier, P.-E.; Chaudet, H.; Delerce, J.; Giraud-Gatineau, A.; Houhamdi, L.; Andrieu, C.; Brechard, L.; Bedotto, M.; Prudent, E.; et al. Analysis of SARS-CoV-2 Variants From 24,181 Patients Exemplifies the Role of Globalization and Zoonosis in Pandemics. Front. Microbiol. 2021, 12, 786233. [Google Scholar] [CrossRef]
- World Health Organization. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 16 September 2024).
- Wilkinson, E.; Giovanetti, M.; Tegally, H.; San, J.E.; Lessells, R.; Cuadros, D.; Martin, D.P.; Rasmussen, D.A.; Zekri, A.-R.N.; Sangare, A.K.; et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science 2021, 374, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Hossain, M.J.; Nahar, Z.; Shahriar, M.; Bhuiyan, M.A.; Islam, M.R. Emerging SARS-CoV-2 Variants and Subvariants: Challenges and Opportunities in the Context of COVID-19 Pandemic. Environ. Health Insights 2022, 16, 11786302221129396. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Frydman, J.; Andino, R. The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 2013, 11, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, R.; Gaonkar, S.L.; Saleh, E.A.M.; Husain, K. A comprehensive review on Covid-19 Omicron (B.1.1.529) variant. Saudi J. Biol. Sci. 2022, 29, 103372. [Google Scholar] [CrossRef] [PubMed]
- Farahat, R.A.; Baklola, M.; Umar, T.P. Omicron B.1.1.529 subvariant: Brief evidence and future prospects. Ann. Med. Surg. 2022, 83, 104808. [Google Scholar] [CrossRef]
- Farahat, R.A.; Abdelaal, A.; Umar, T.P.; El-Sakka, A.A.; Benmelouka, A.Y.; Albakri, K.; Ali, I.; Al-Ahdal, T.; Abdelazeem, B.; Sah, R.; et al. The emergence of SARS-CoV-2 Omicron subvariants: Current situation and future trends. InfezMed 2022, 30, 480–494. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Grayo, S.; Troupin, C.; Diagne, M.M.; Sagno, H.; Ellis, I.; Doukouré, B.; Diallo, A.; Bart, J.-M.; Kaba, M.L.; Henry, B.; et al. SARS-CoV-2 Circulation, Guinea, March 2020–July 2021. Emerg. Infect. Dis. 2022, 28, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.; Hadfield, J.; Sibley, T.R.; Lee, J.; Fay, K.; Ilcisin, M.; Harkins, E.; Bedford, T.; Neher, R.A.; Hodcroft, E.B. Augur: A bioinformatics toolkit for phylogenetic analyses of human pathogens. J. Open Source Softw. 2021, 6, 2906. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- AFROSCREEN: Renforcement des Capacités de Séquençage en Afrique. Available online: https://www.afroscreen.org/ (accessed on 26 September 2024).
- Sow, M.S.; Togo, J.; Simons, L.M.; Diallo, S.T.; Magassouba, M.L.; Keita, M.B.; Somboro, A.M.; Coulibaly, Y.; Ozer, E.A.; Hultquist, J.F.; et al. Genomic characterization of SARS-CoV-2 in Guinea, West Africa. PLoS ONE 2024, 19, e0299082. [Google Scholar] [CrossRef] [PubMed]
- Kolié, D.; Keita, F.N.; Delamou, A.; Dossou, J.-P.; Van Damme, W.; Agyepong, I.A. Learning from the COVID-19 pandemic for future epidemics and pandemics preparedness and response in Guinea: Findings from a scoping review. Front. Public Health 2022, 10, 879850. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; Veinotte, K.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, B.; Tang, T.; Daniel, S.; Jaimes, J.A.; Whittaker, G. Functional evaluation of the P681H mutation on the proteolytic activation the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. iScience 2022, 25, 103589. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Anoh, E.A.; Wayoro, O.; Monemo, P.; Belarbi, E.; Sachse, A.; Wilkinson, E.; San, J.E.; Leendertz, F.H.; Diané, B.; Calvignac-Spencer, S.; et al. Subregional origins of emerging SARS-CoV-2 variants during the second pandemic wave in Côte d’Ivoire. Virus Genes 2023, 59, 370–376. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnimadi, T.A.C.; Kadio, K.J.-J.O.; Mathew, M.J.; Diallo, H.; Soumah, A.K.; Keita, A.K.; Hounmenou, C.G.; Fernandez-Nuñez, N.; Vidal, N.; Guichet, E.; et al. Genetic Diversity and Spatiotemporal Distribution of SARS-CoV-2 Variants in Guinea: A Meta-Analysis of Sequence Data (2020–2023). Viruses 2025, 17, 204. https://doi.org/10.3390/v17020204
Gnimadi TAC, Kadio KJ-JO, Mathew MJ, Diallo H, Soumah AK, Keita AK, Hounmenou CG, Fernandez-Nuñez N, Vidal N, Guichet E, et al. Genetic Diversity and Spatiotemporal Distribution of SARS-CoV-2 Variants in Guinea: A Meta-Analysis of Sequence Data (2020–2023). Viruses. 2025; 17(2):204. https://doi.org/10.3390/v17020204
Chicago/Turabian StyleGnimadi, Thibaut Armel Chérif, Kadio Jean-Jacques Olivier Kadio, Mano Joseph Mathew, Haby Diallo, Abdoul Karim Soumah, Alpha Kabiné Keita, Castro Gbêmêmali Hounmenou, Nicolas Fernandez-Nuñez, Nicole Vidal, Emilande Guichet, and et al. 2025. "Genetic Diversity and Spatiotemporal Distribution of SARS-CoV-2 Variants in Guinea: A Meta-Analysis of Sequence Data (2020–2023)" Viruses 17, no. 2: 204. https://doi.org/10.3390/v17020204
APA StyleGnimadi, T. A. C., Kadio, K. J.-J. O., Mathew, M. J., Diallo, H., Soumah, A. K., Keita, A. K., Hounmenou, C. G., Fernandez-Nuñez, N., Vidal, N., Guichet, E., Ayouba, A., Delaporte, E., Peeters, M., Touré, A., & Keita, A. K. (2025). Genetic Diversity and Spatiotemporal Distribution of SARS-CoV-2 Variants in Guinea: A Meta-Analysis of Sequence Data (2020–2023). Viruses, 17(2), 204. https://doi.org/10.3390/v17020204