
Modulation of Ire1-Xbp1 Defense Pathway in Encephalomyocarditis Virus-Infected HeLa Cells
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Western Blot Analysis of HeLa Cell Lysates
2.3. RNA Extraction and RT-qPCR
2.4. PCR Experiment for Differentiation Spliced and Unspliced XBP1 mRNA Forms
2.5. Western Blot Quantification Analysis
3. Results
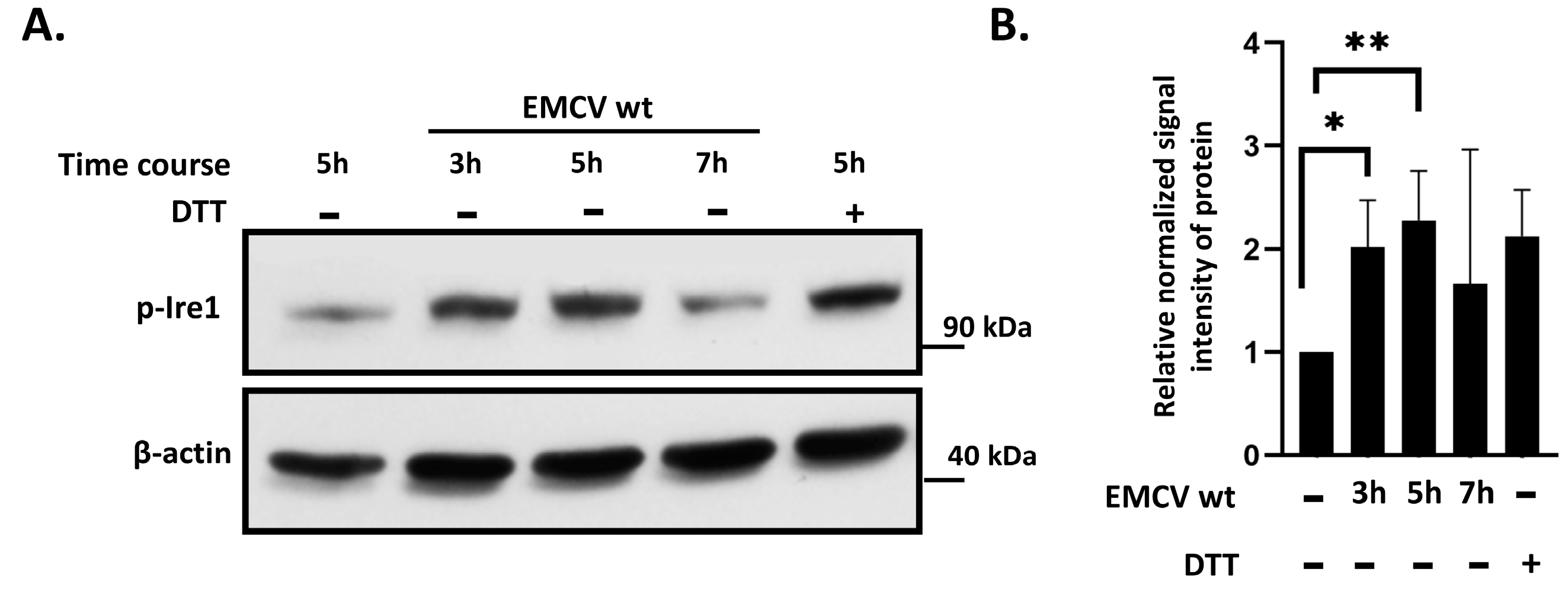
3.1. IRE1 Phosphorylation Is Activated in EMCV-Infected HeLa Cells
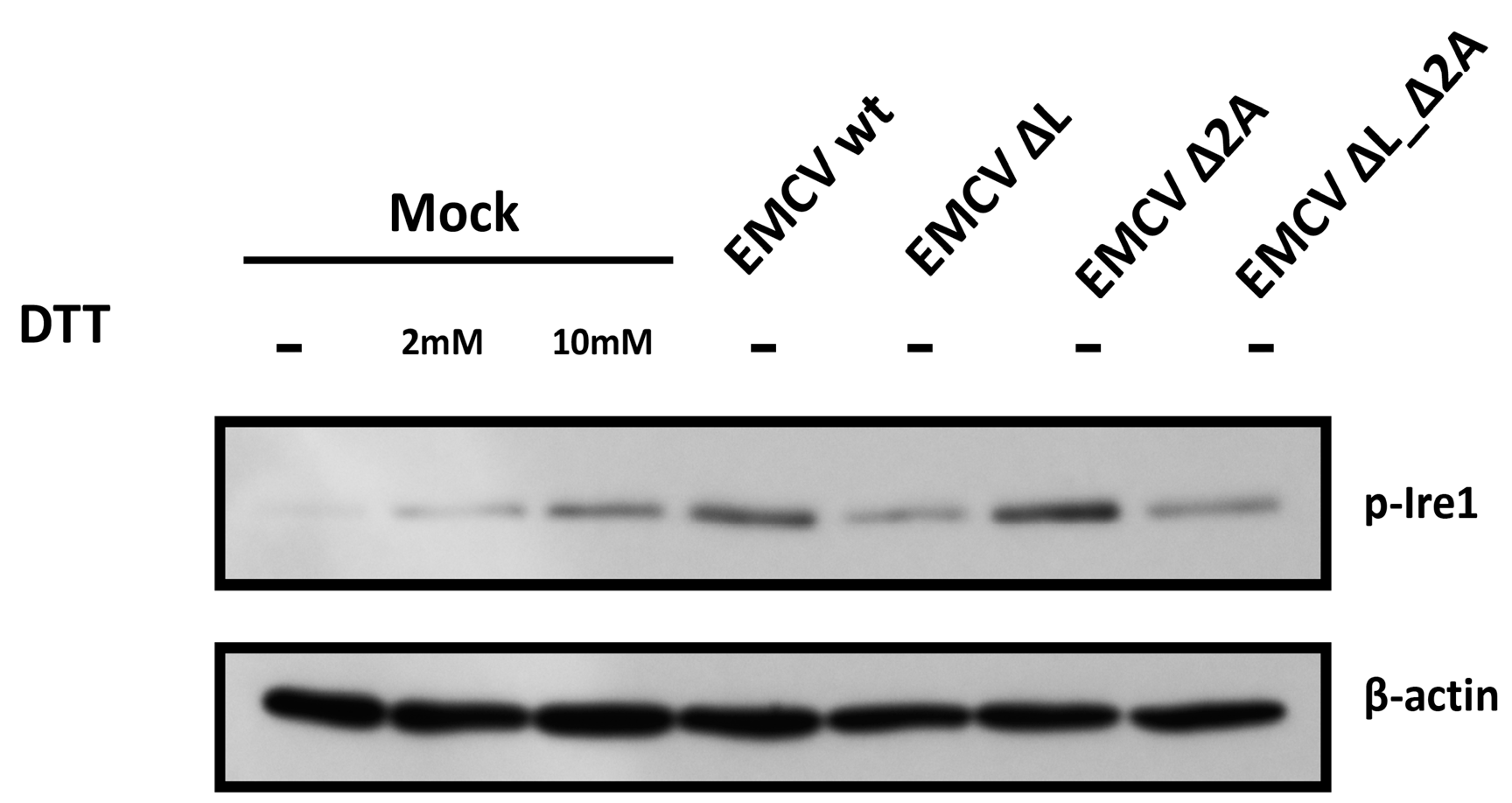
3.2. IRE1 Phosphorylation Levels in HeLa Cells Infected with EMCV Deficient in L or 2A Protein
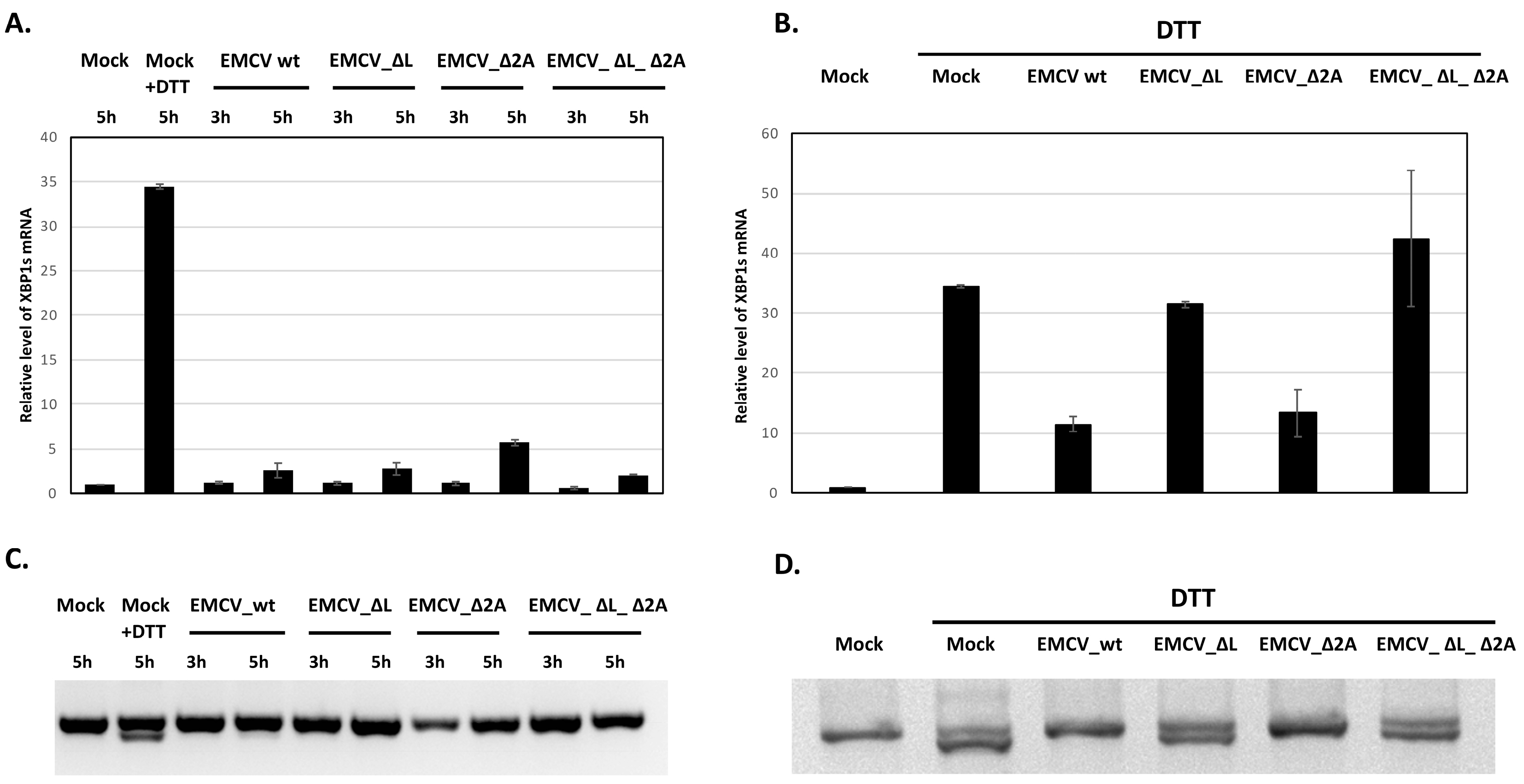
3.3. Inhibition of XBP1 mRNA Splicing in EMCV-Infected HeLa Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carocci, M.; Bakkali-Kassimi, L. The Encephalomyocarditis Virus. Virulence 2012, 3, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Jiang, K.; Zhang, G.; Wang, X.; Li, Y.; Jiang, P. Pathogenicity and Molecular Analysis of an Encephalomyocarditis Virus Isolate from Mideastern China. Can. J. Vet. Res. 2012, 76, 157–160. [Google Scholar]
- Feng, R.; Wei, J.; Zhang, H.; Fan, J.; Li, X.; Wang, D.; Xie, J.; Qiao, Z.; Li, M.; Bai, J.; et al. National Serosurvey of Encephalomyocarditis Virus in Healthy People and Pigs in China. Arch. Virol. 2015, 160, 2957–2964. [Google Scholar] [CrossRef]
- Seaman, J.T.; Boulton, J.G.; Carrigan, M.J. Encephalomyocarditis Virus Disease of Pigs Associated with a Plague of Rodents. Aust. Vet. J. 1986, 63, 292–294. [Google Scholar] [CrossRef]
- Bardina, M.V.; Lidsky, P.V.; Sheval, E.V.; Fominykh, K.V.; Van Kuppeveld, F.J.M.; Polyakov, V.Y.; Agol, V.I. Mengovirus-Induced Rearrangement of the Nuclear Pore Complex: Hijacking Cellular Phosphorylation Machinery. J. Virol. 2009, 83, 3150–3161. [Google Scholar] [CrossRef] [PubMed]
- Lidsky, P.V.; Hato, S.; Bardina, M.V.; Aminev, A.G.; Palmenberg, A.C.; Sheval, E.V.; Polyakov, V.Y.; Van Kuppeveld, F.J.M.; Agol, V.I. Nucleocytoplasmic Traffic Disorder Induced by Cardioviruses. J. Virol. 2006, 80, 2705–2717. [Google Scholar] [CrossRef]
- Cornilescu, C.C.; Porter, F.W.; Zhao, K.Q.; Palmenberg, A.C.; Markley, J.L. NMR Structure of the Mengovirus Leader Protein Zinc-finger Domain. FEBS Lett. 2008, 582, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, C.M.T.; Hall, D.J.; Hill, M.; Riddle, M.; Pranter, A.; Dillman, J.; Deibel, M.; Palmenberg, A.C. Leader Protein of Encephalomyocarditis Virus Binds Zinc, Is Phosphorylated during Viral Infection, and Affects the Efficiency of Genome Translation. Virology 2001, 290, 261–271. [Google Scholar] [CrossRef]
- Bacot-Davis, V.R.; Ciomperlik, J.J.; Basta, H.A.; Cornilescu, C.C.; Palmenberg, A.C. Solution Structures of Mengovirus Leader Protein, Its Phosphorylated Derivatives, and in Complex with Nuclear Transport Regulatory Protein, RanGTPase. Proc. Natl. Acad. Sci. USA 2014, 111, 15792–15797. [Google Scholar] [CrossRef]
- Ivin, Y.Y.; Butusova, A.A.; Gladneva, E.E.; Kolomijtseva, G.Y.; Khapchaev, Y.K.; Ishmukhametov, A.A. The role of the encephalomyocarditis virus type 1 proteins L and 2A in the inhibition of the synthesis of cellular proteins and the accumulation of viral proteins during infection. Probl. Virol. 2023, 68, 428–444. [Google Scholar] [CrossRef]
- Groppo, R.; Brown, B.A.; Palmenberg, A.C. Mutational Analysis of the EMCV 2A Protein Identifies a Nuclear Localization Signal and an eIF4E Binding Site. Virology 2011, 410, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Liang, L.; Qin, T.; Xiao, S.; Liang, R. Encephalomyocarditis Virus 2A Protein Inhibited Apoptosis by Interaction with Annexin A2 through JNK/c-Jun Pathway. Viruses 2022, 14, 359. [Google Scholar] [CrossRef] [PubMed]
- Zoll, J.; Galama, J.M.; Van Kuppeveld, F.J.; Melchers, W.J. Mengovirus Leader Is Involved in the Inhibition of Host Cell Protein Synthesis. J. Virol. 1996, 70, 4948–4952. [Google Scholar] [CrossRef]
- Ivin, Y.; Butusova, A.; Gladneva, E.; Gmyl, A.; Ishmukhametov, A. Comprehensive Elucidation of the Role of L and 2A Security Proteins on Cell Death during EMCV Infection. Viruses 2024, 16, 280. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein Translation and Folding Are Coupled by an Endoplasmic-Reticulum-Resident Kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the Mechanisms of Apoptosis Induced by Endoplasmic Reticulum Stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases That Process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Bashir, S.; Banday, M.; Qadri, O.; Bashir, A.; Hilal, N.; Nida-i-Fatima; Rader, S.; Fazili, K.M. The Molecular Mechanism and Functional Diversity of UPR Signaling Sensor IRE1. Life Sci. 2021, 265, 118740. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.; Wang, A. Divergence and Conservation of the Major UPR Branch IRE1-bZIP Signaling Pathway across Eukaryotes. Sci. Rep. 2016, 6, 27362. [Google Scholar] [CrossRef]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian Endoplasmic Reticulum Stress Sensor IRE1 Signals by Dynamic Clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Kang, T.-I.; So, J.-S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Bright, M.D.; Itzhak, D.N.; Wardell, C.P.; Morgan, G.J.; Davies, F.E. Cleavage of BLOC1S1 mRNA by IRE1 Is Sequence Specific, Temporally Separate from XBP1 Splicing, and Dispensable for Cell Viability under Acute Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2015, 35, 2186–2202. [Google Scholar] [CrossRef]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 Has Distinct Catalytic Mechanisms for XBP1/HAC1 Splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER Stress Sensor and Cell Fate Executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef]
- Zhang, H.M.; Ye, X.; Su, Y.; Yuan, J.; Liu, Z.; Stein, D.A.; Yang, D. Coxsackievirus B3 Infection Activates the Unfolded Protein Response and Induces Apoptosis through Downregulation of P58 IPK and Activation of CHOP and SREBP1. J. Virol. 2010, 84, 8446–8459. [Google Scholar] [CrossRef]
- Jheng, J.-R.; Lau, K.-S.; Lan, Y.-W.; Horng, J.-T. A Novel Role of ER Stress Signal Transducer ATF6 in Regulating Enterovirus A71 Viral Protein Stability. J. Biomed. Sci. 2018, 25, 9. [Google Scholar] [CrossRef]
- Song, J.; Chi, M.; Luo, X.; Song, Q.; Xia, D.; Shi, B.; Han, J. Non-Structural Protein 2B of Human Rhinovirus 16 Activates Both PERK and ATF6 Rather Than IRE1 to Trigger ER Stress. Viruses 2019, 11, 133. [Google Scholar] [CrossRef]
- Hou, L.; Ge, X.; Xin, L.; Zhou, L.; Guo, X.; Yang, H. Nonstructural Proteins 2C and 3D Are Involved in Autophagy as Induced by the Encephalomyocarditis Virus. Virol. J. 2014, 11, 156. [Google Scholar] [CrossRef]
- Xie, J.; Idris, A.; Feng, R. The Complex Interplay between Encephalomyocarditis Virus and the Host Defence System. Virulence 2024, 15, 2383559. [Google Scholar] [CrossRef] [PubMed]
- Van Der Grein, S.G.; Defourny, K.A.Y.; Rabouw, H.H.; Goerdayal, S.S.; Van Herwijnen, M.J.C.; Wubbolts, R.W.; Altelaar, M.; Van Kuppeveld, F.J.M.; Nolte-‘t Hoen, E.N.M. The Encephalomyocarditis Virus Leader Promotes the Release of Virions inside Extracellular Vesicles via the Induction of Secretory Autophagy. Nat. Commun. 2022, 13, 3625. [Google Scholar] [CrossRef] [PubMed]
- Shishova, A.; Dyugay, I.; Fominykh, K.; Baryshnikova, V.; Dereventsova, A.; Turchenko, Y.; Slavokhotova, A.A.; Ivin, Y.; Dmitriev, S.E.; Gmyl, A. Enteroviruses Manipulate the Unfolded Protein Response through Multifaceted Deregulation of the Ire1-Xbp1 Pathway. Viruses 2022, 14, 2486. [Google Scholar] [CrossRef]
- Wells, K.M.; He, K.; Pandey, A.; Cabello, A.; Zhang, D.; Yang, J.; Gomez, G.; Liu, Y.; Chang, H.; Li, X.; et al. Brucella Activates the Host RIDD Pathway to Subvert BLOS1-Directed Immune Defense. eLife 2022, 11, e73625. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Sen, U.; Vrati, S. Regulated IRE1-Dependent Decay Pathway Is Activated during Japanese Encephalitis Virusinduced Unfolded Protein Response and Benefits Viral Replication. J. Gen. Virol. 2014, 95, 71–79. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shishova, A.; Ivin, Y.; Gladneva, E.; Fominykh, K.; Dyugay, I.; Gmyl, A. Modulation of Ire1-Xbp1 Defense Pathway in Encephalomyocarditis Virus-Infected HeLa Cells. Viruses 2025, 17, 360. https://doi.org/10.3390/v17030360
Shishova A, Ivin Y, Gladneva E, Fominykh K, Dyugay I, Gmyl A. Modulation of Ire1-Xbp1 Defense Pathway in Encephalomyocarditis Virus-Infected HeLa Cells. Viruses. 2025; 17(3):360. https://doi.org/10.3390/v17030360
Chicago/Turabian StyleShishova, Anna, Yury Ivin, Ekaterina Gladneva, Ksenia Fominykh, Ilya Dyugay, and Anatoly Gmyl. 2025. "Modulation of Ire1-Xbp1 Defense Pathway in Encephalomyocarditis Virus-Infected HeLa Cells" Viruses 17, no. 3: 360. https://doi.org/10.3390/v17030360
APA StyleShishova, A., Ivin, Y., Gladneva, E., Fominykh, K., Dyugay, I., & Gmyl, A. (2025). Modulation of Ire1-Xbp1 Defense Pathway in Encephalomyocarditis Virus-Infected HeLa Cells. Viruses, 17(3), 360. https://doi.org/10.3390/v17030360