The Role of Combined Penetration Enhancers in Nasal Microspheres on In Vivo Drug Bioavailability

 ,
, 
 and
and
Abstract
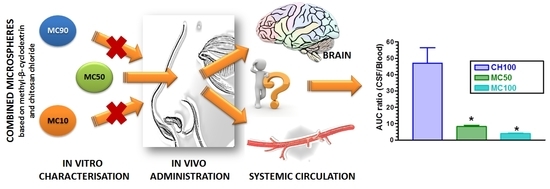
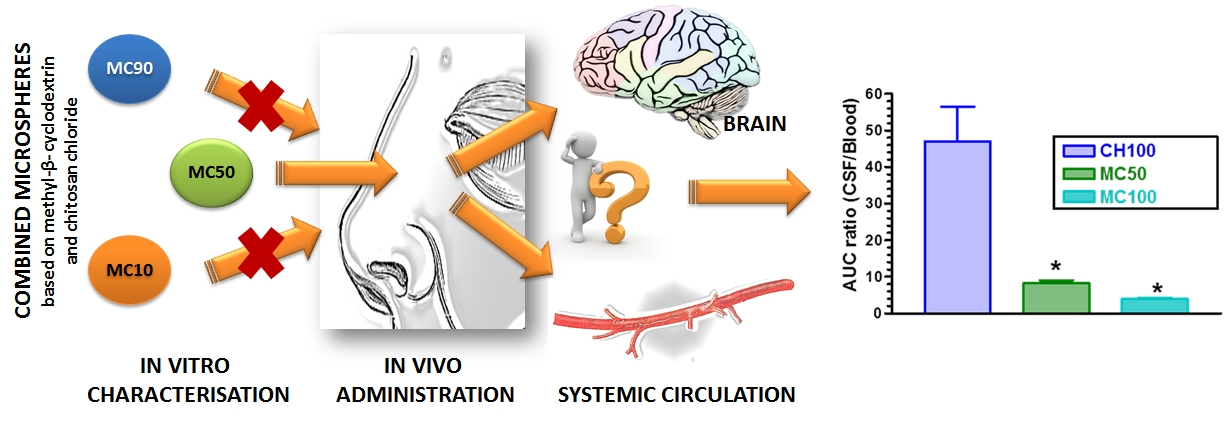
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Microspheres
2.3. Microsphere Characterization
2.3.1. Determination of Drug Content and Encapsulation Efficiency
2.3.2. Morphological Analysis
2.3.3. Particle Size Analysis
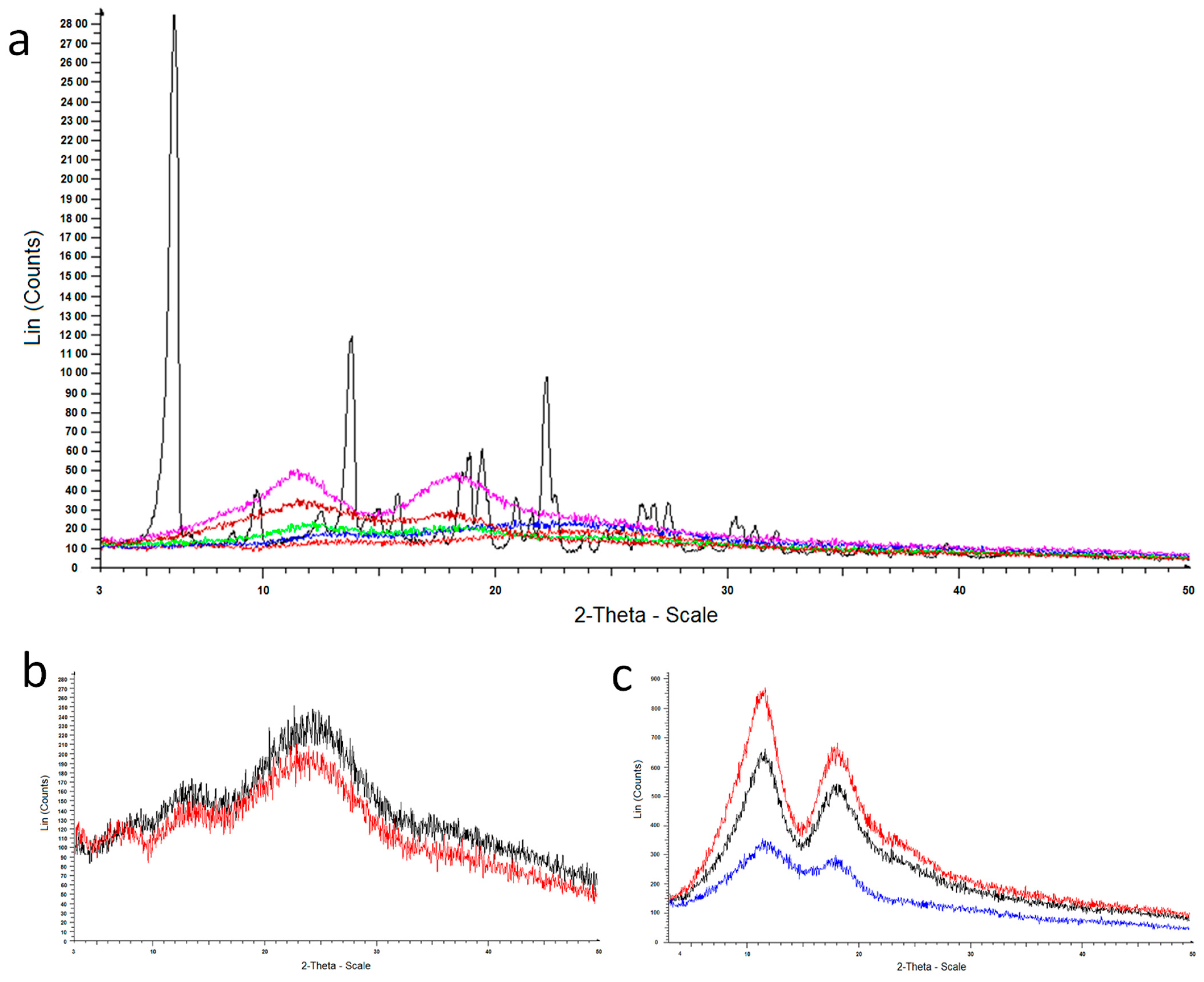
2.3.4. Powder X-ray Diffraction
2.3.5. Water Uptake
2.3.6. In Vitro Permeation Test of Microspheres
2.4. In Vivo Studies
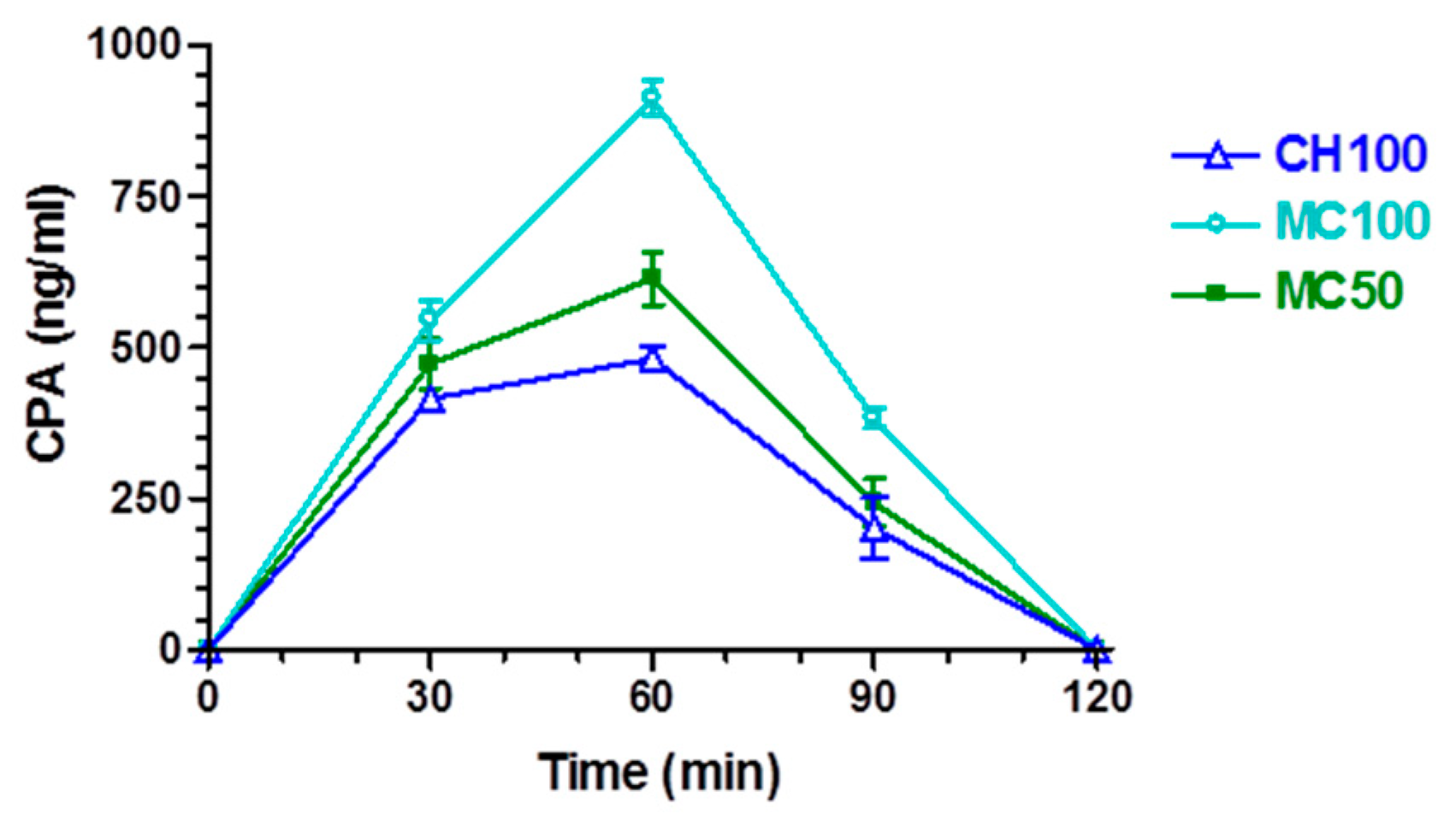
2.4.1. In Vivo CPA Administration and Quantification
2.4.2. HPLC Analysis
2.5. Statistical Analysis
3. Results and Discussion
3.1. Preparation and Characterization of Microspheres
3.1.1. Determination of Drug Content and Encapsulation Efficiency
3.1.2. Morphological Analysis
3.1.3. Particle Size Analysis
3.1.4. Powder X-ray Diffraction
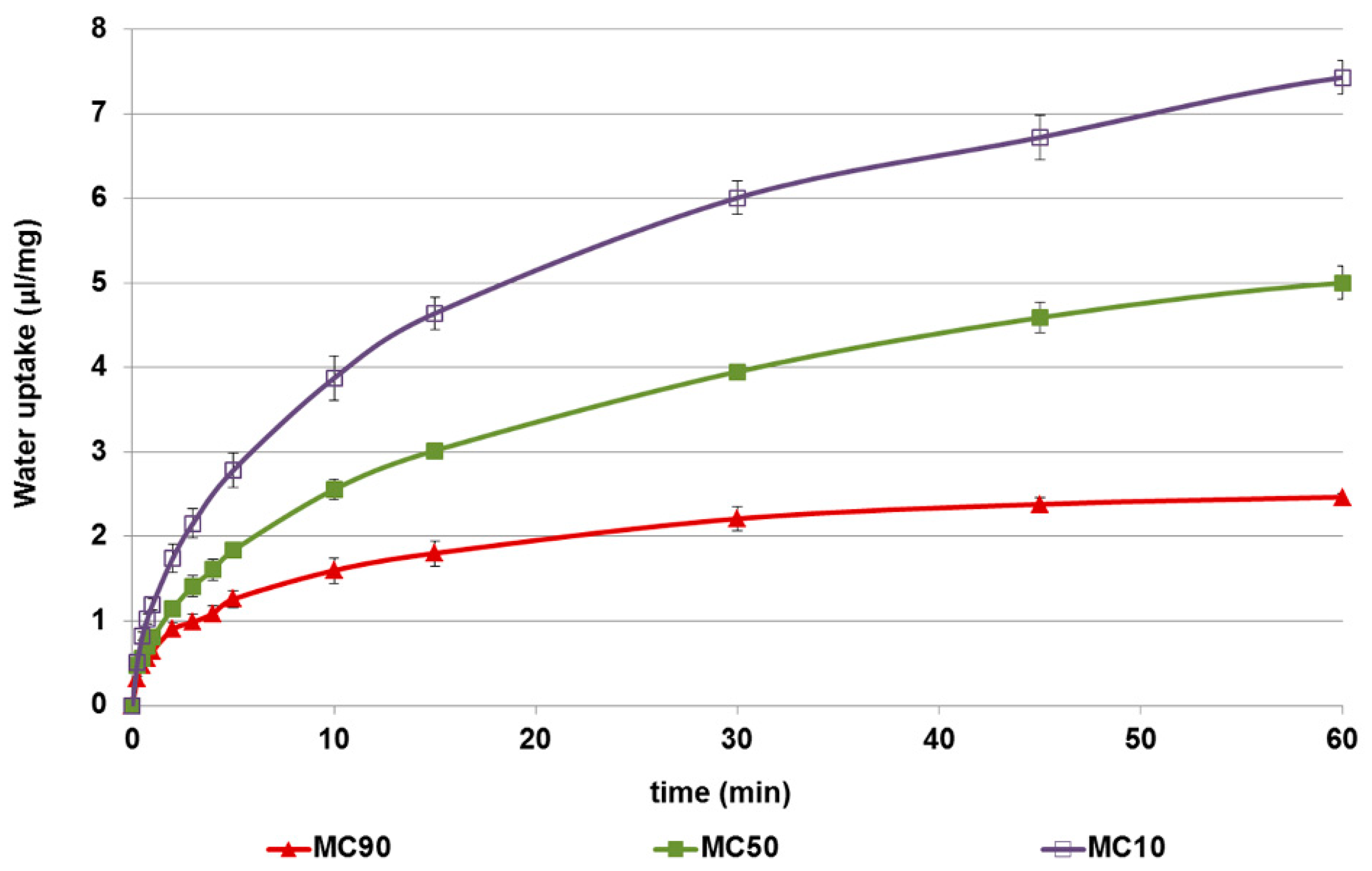
3.1.5. Water Uptake
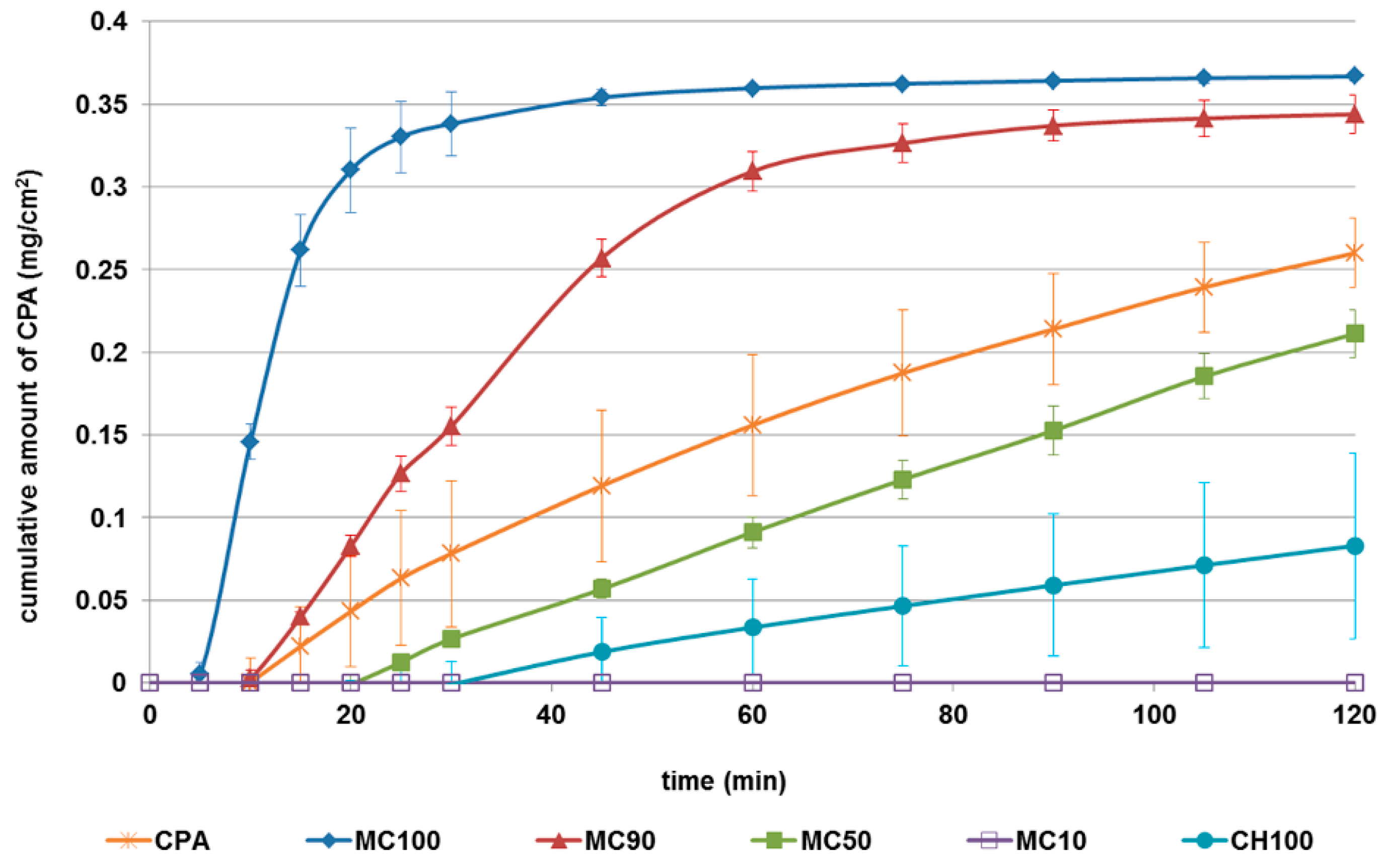
3.1.6. In Vitro Permeation Test of Microspheres
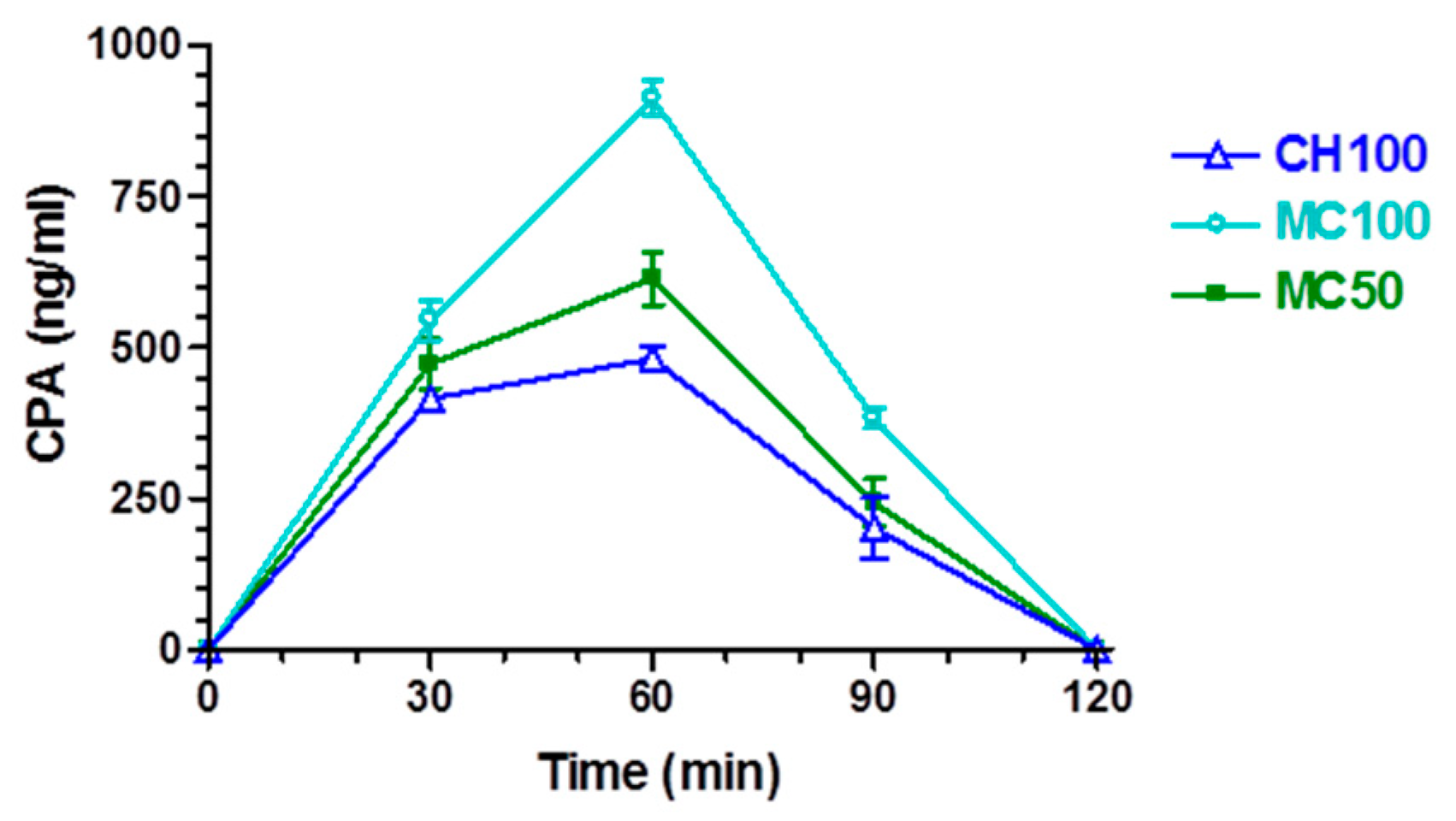
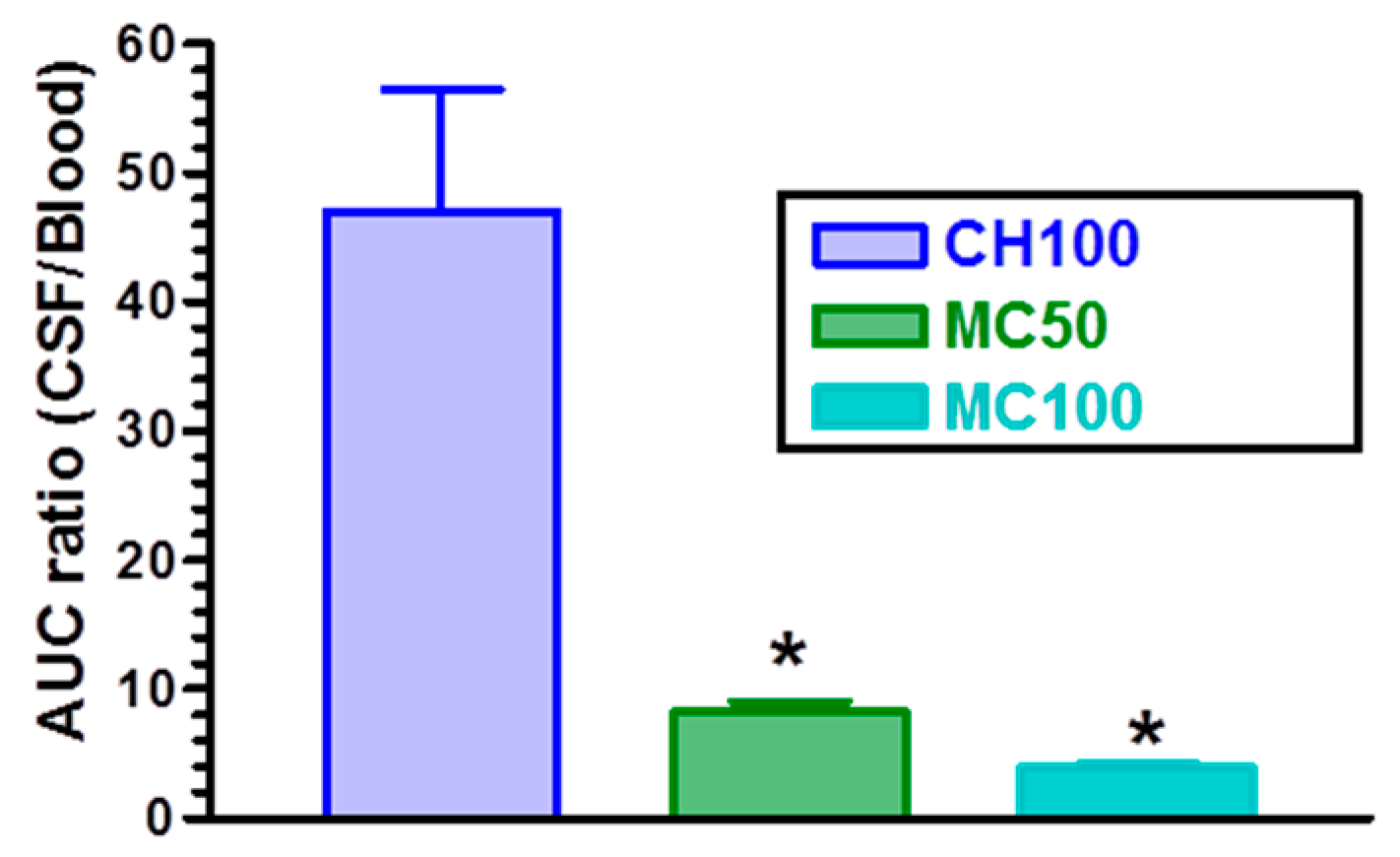
3.2. In Vivo CPA Administration and Quantification
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aungst, B.J. Absorption Enhancers: Applications and Advances. AAPS J. 2012, 14, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.S.; Illum, L. Absorption Enhancers for Nasal Drug Delivery. Clin. Pharmacokinet. 2003, 42, 1107–1128. [Google Scholar] [CrossRef] [PubMed]
- Gavini, E.; Rassu, G.; Haukvik, T.; Lanni, C.; Racchi, M.; Giunchedi, P. Mucoadhesive microspheres for nasal administration of cyclodextrins. J. Drug Target. 2009, 17, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Gavini, E.; Rassu, G.; Ferraro, L.; Beggiato, S.; Alhalaweh, A.; Velaga, S.; Marchetti, N.; Bandiera, P.; Giunchedi, P.; Dalpiaz, A. Influence of polymeric microcarriers on the in vivo intranasal uptake of an anti-migraine drug for brain targeting. Eur. J. Pharm. Biopharm. 2013, 83, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, A.; Soddu, E.; Turunc Bayrakdar, E.; Uyanikgil, Y.; Kanit, L.; Armagan, G.; Rassu, G.; Gavini, E.; Giunchedi, P. Neuroprotective Effects of Engineered Polymeric Nasal Microspheres Containing Hydroxypropyl-β-cyclodextrin on β-Amyloid (1-42)-Induced Toxicity. J. Pharm. Sci. 2016, 105, 2372–2380. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.C.; Barry, B.W. Penetration enhancers. Adv. Drug Deliv. Rev. 2012, 64, 128–137. [Google Scholar] [CrossRef]
- Illum, L. Nasal drug delivery-possibilities, problems and solutions. J. Control. Release 2003, 87, 187–198. [Google Scholar] [CrossRef]
- Merkus, F.W.H.M.; Verhoef, J.C.; Marttin, E.; Romeijn, S.G.; van der Kuy, P.H.M.; Hermens, W.A.J.; Schipper, N.G.M. Cyclodextrins in nasal drug delivery. Adv. Drug Deliv. Rev. 1999, 36, 41–57. [Google Scholar] [CrossRef]
- Yang, T.; Hussain, A.; Paulson, J.; Abbruscato, T.J.; Ahsan, F. Cyclodextrins in nasal delivery of low-molecular-weight heparins: In vivo and in vitro studies. Pharm. Res. 2004, 21, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, X.; Wu, Z.; Gao, X.; Shua, S.; Wang, Z.; Li, C. β-Cyclodextrin grafting hyperbranched polyglycerols as carriers for nasal insulin delivery. Carbohydr. Polym. 2011, 84, 1419–1425. [Google Scholar] [CrossRef]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in Drug Delivery: An Updated Review. AAPS PharmSciTech 2005, 6, E329–E357. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.; O’Neill, C.A.; Padfield, P.J. Depletion of Caco-2 cell cholesterol disrupts barrier function by altering the detergent solubility and distribution of specific tight-junction proteins. Biochem. J. 2005, 387, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Fenyvesi, F.; Réti-Nagy, K.; Bacsó, Z.; Gutay-Tóth, Z.; Malanga, M.; Fenyvesi, É.; Szente, L.; Váradi, J.; Ujhelyi, Z.; Fehér, P.; et al. Fluorescently labeled methyl-beta-cyclodextrin enters intestinal epithelial Caco-2 cells by fluid-phase endocytosis. PLoS ONE 2014, 9, e84856. [Google Scholar] [CrossRef] [PubMed]
- Casettari, L.; Illum, L. Chitosan in nasal delivery systems for therapeutic drugs. J. Control. Release 2014, 190, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Gavini, E.; Rassu, G.; Ferraro, L.; Generosi, A.; Rau, J.V.; Brunetti, A.; Giunchedi, P.; Dalpiaz, A. Influence of chitosan glutamate on the in vivo intranasal absorption of rokitamycin from microspheres. J. Pharm. Sci. 2011, 100, 1488–1502. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Fogagnolo, M.; Ferraro, L.; Capuzzo, A.; Pavan, B.; Rassu, G.; Salis, A.; Giunchedi, P.; Gavini, E. Nasal chitosan microparticles target a zidovudine prodrug to brain HIV sanctuaries. Antiviral Res. 2015, 123, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Rassu, G.; Soddu, E.; Cossu, M.; Gavini, E.; Giunchedi, P.; Dalpiaz, A. Particulate formulations based on chitosan for nose-to-brain delivery of drugs. A review. J. Drug Deliv. Sci. Technol. 2016, 32, 77–87. [Google Scholar] [CrossRef]
- Council of Europe. European Pharmacopoeia Chitosan Hydrochloride, 8th ed.; Council of Europe: Strasbourg, France, 2014; p. 1841. [Google Scholar]
- Türker, S.; Onur, E.; Ozer, Y. Nasal route and drug delivery systems. Pharm. World Sci. 2004, 26, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Rassu, G.; Soddu, E.; Cossu, M.; Brundu, A.; Cerri, G.; Marchetti, N.; Ferraro, L.; Regan, R.F.; Giunchedi, P.; Gavini, E.; et al. Solid microparticles based on chitosan or methyl-β-cyclodextrin: A first formulative approach to increase the nose-to-brain transport of deferoxamine mesylate. J. Control. Release 2015, 201, 68–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bshara, H.; Osman, R.; Mansour, S.; El-Shamy, A.E.A. Chitosan and cyclodextrin in intranasal microemulsion for improved brain buspirone hydrochloride pharmacokinetics in rats. Carbohydr. Polym. 2014, 99, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Gavini, E.; Colombo, G.; Russo, P.; Bortolotti, F.; Ferraro, L.; Tanganelli, S.; Scatturin, A.; Menegatti, E.; Giunchedi, P. Brain uptake of an anti-ischemic agent by nasal administration of microparticles. J. Pharm. Sci. 2008, 97, 4889–4903. [Google Scholar] [CrossRef] [PubMed]
- Gavini, E.; Rassu, G.; Ciarnelli, V.; Spada, G.; Cossu, M.; Giunchedi, P. Mucoadhesive drug delivery systems for nose-to-brain targeting of dopamine. J. Nanoneurosci. 2012, 2, 1–9. [Google Scholar] [CrossRef]
- Rassu, G.; Gavini, E.; Jonassen, H.; Zambito, Y.; Fogli, S.; Breschi, M.C.; Giunchedi, P. New chitosan derivatives for the preparation of rokitamycin loaded microspheres designed for ocular or nasal administration. J. Pharm. Sci. 2009, 98, 4852–4865. [Google Scholar] [CrossRef] [PubMed]
- Gavini, E.; Spada, G.; Rassu, G.; Cerri, G.; Brundu, A.; Cossu, M.; Sorrenti, M.; Giunchedi, P. Development of solid nanoparticles based on hydroxypropyl-β-cyclodextrin aimed for the colonic transmucosal delivery of diclofenac sodium. J. Pharm. Pharmacol. 2011, 63, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Kouchak, M.; Handali, S.; Boroujeni, B.N. Evaluation of the mechanical properties and drug permeability of chitosan/Eudragit RL composite film. Osong Public Health Res. Perspect. 2015, 6, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, M.P.; Romeijn, S.G.; Verhoef, J.C.; Merkus, F.W. Serial cerebrospinal fluid sampling in a rat model to study drug uptake from the nasal cavity. J. Neurosci. Methods 2002, 116, 99–107. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Ferraro, L.; Perrone, D.; Leo, E.; Iannuccelli, V.; Pavan, B.; Paganetto, G.; Beggiato, S.; Scalia, S. Brain uptake of a Zidovudine prodrug after nasal administration of solid lipid microparticles. Mol. Pharm. 2014, 11, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, C.; Artusi, M.; Santi, P.; Colombo, P. Caffeine microparticles for nasal administration obtained by spray drying. Int. J. Pharm. 2002, 242, 335–339. [Google Scholar] [CrossRef]
- Felgenhauer, K. Protein Size and CSF Composition. Klinische Wochensch. 1974, 52, 1158–1164. [Google Scholar] [CrossRef]
- Madu, A.; Cioffe, C.; Mian, U.; Burroughs, M.; Tuomanen, E.; Mayers, M.; Schwartz, E.; Miller, M. Pharmacokinetics of Fluconazole in Cerebrospinal Fluid and Serum of Rabbits: Validation of an Animal Model used to Measure Drug Concentrations in Cerebrospinal Fluid. Antimicrob. Agents Chemother. 1994, 38, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Gavini, E.; Rassu, G.; Sanna, V.; Cossu, M.; Giunchedi, P. Mucoadhesive microspheres for nasal administration of an antiemetic drug, metoclopramide: In-vitro/ex-vivo studies. J. Pharm. Pharmacol. 2005, 57, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Mathot, R.A.; Appel, S.; van Schaick, E.A.; Soudijn, W.; Ijzerman, A.P.; Danhof, M. High-performance liquid chromatography of the adenosine A1 agonist N6-cyclopentyladenosine and the A1 antagonist 8-cyclopentyltheophylline and its application in a pharmacokinetic study in rats. J. Chromatogr. 1993, 620, 113–120. [Google Scholar] [CrossRef]
- Brodie, M.S.; Lee, K.; Fredholm, B.B.; Stahle, L.; Dunwiddie, T.V. Central versus peripheral mediation of responses to adenosine receptor agonists: Evidence against a central mode of action. Brain Res. 1987, 415, 323–330. [Google Scholar] [CrossRef]
- Tamai, I.; Tsuji, A. Transporter-mediated permeation of drugs across the blood-brain barrier. J. Pharm. Sci. 2000, 89, 1371–1388. [Google Scholar] [CrossRef]
- Schaddelee, M.P.; Read, K.D.; Cleypool, C.G.J.; Ijzerman, A.P.; Danhof, M.; de Boer, A.G. Brain penetration of synthetic A1 receptor agonists in situ: Role of the rENT1 nucleoside transporter and binding to blood constituents. Eur. J. Pharm. Sci. 2005, 24, 59–66. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microsphere Codes | Methyl-β-Cyclodextrin | Chitosan | DC (%) | EE (%) | dvs (µm) | SPAN |
|---|---|---|---|---|---|---|
| MC100 | 100 | - | 4.25 ± 0.05 a | 88.78 ± 0.98 a | 3.49 ± 0.10 | 2.20 |
| MC90 | 90 | 10 | 4.25 ± 0.01 a | 88.66 ± 0.22 a | 2.74 ± 0.09 | 1.85 |
| MC50 | 50 | 50 | 4.20 ± 0.07 a | 87.87 ± 1.53 a | 6.79 ± 1.84 b,c | 2.24 |
| MC10 | 10 | 90 | 3.81 ± 0.05 | 80.91 ± 1.13 | 6.55 ± 1.04 b,c | 2.09 |
| CH100 # | - | 100 | 4.75 ± 0.06 | 95.0 ± 1.20 | 6.14 ± 0.84 b,c | 1.66 |
| Microsphere Codes | J (μg/cm2 min) | T Lag (min) | Drug Permeated (%) |
|---|---|---|---|
| CPA | 2.48 ± 0.65 a,b,c | 1.44 | 83.55 ± 10.85 d,e,g |
| MC100 | 25.64 ± 2.14 a | 4.64 | 112.56 ± 1.24 d |
| MC90 | 7.27 ± 0.18 a,b | 8.96 | 105.87 ± 2.26 e |
| MC50 | 2.09 ± 0.14 a,b | 17.62 | 65.46 ± 3.66 d,e,f |
| MC10 | 0.00 a,b,c | - | 0.00 |
| CH100 | 0.86 ± 0.53 a,b | 20.11 | 22.26 ± 15.33 d,e,f,g |
| Blood | CSF | |||
|---|---|---|---|---|
| Administration Way | AUC (ng mL−1 min) | Absolute Bioavailability | AUC (ng mL−1 min) | Relative Bioavailability |
| Intravenous | 37,687 ± 1118 | 100 | n.d. | 0 |
| Nasal (suspension) | n.d. | 0 | n.d. | 0 |
| Nasal (CH100) | 699 ± 257 | 1.85% | 32,910 ± 1199 | 100% |
| Nasal (MC50) | 4830 ± 827 a | 12.8% | 39,900 ± 1520 | 121% b |
| Nasal (MC100) | 13,590 ± 1451 c | 36.0% | 55,140 ± 1016 | 170% c |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rassu, G.; Ferraro, L.; Pavan, B.; Giunchedi, P.; Gavini, E.; Dalpiaz, A. The Role of Combined Penetration Enhancers in Nasal Microspheres on In Vivo Drug Bioavailability. Pharmaceutics 2018, 10, 206. https://doi.org/10.3390/pharmaceutics10040206
Rassu G, Ferraro L, Pavan B, Giunchedi P, Gavini E, Dalpiaz A. The Role of Combined Penetration Enhancers in Nasal Microspheres on In Vivo Drug Bioavailability. Pharmaceutics. 2018; 10(4):206. https://doi.org/10.3390/pharmaceutics10040206
Chicago/Turabian StyleRassu, Giovanna, Luca Ferraro, Barbara Pavan, Paolo Giunchedi, Elisabetta Gavini, and Alessandro Dalpiaz. 2018. "The Role of Combined Penetration Enhancers in Nasal Microspheres on In Vivo Drug Bioavailability" Pharmaceutics 10, no. 4: 206. https://doi.org/10.3390/pharmaceutics10040206
APA StyleRassu, G., Ferraro, L., Pavan, B., Giunchedi, P., Gavini, E., & Dalpiaz, A. (2018). The Role of Combined Penetration Enhancers in Nasal Microspheres on In Vivo Drug Bioavailability. Pharmaceutics, 10(4), 206. https://doi.org/10.3390/pharmaceutics10040206