Prospects and Challenges of Phospholipid-Based Prodrugs

 ,
, 

Abstract
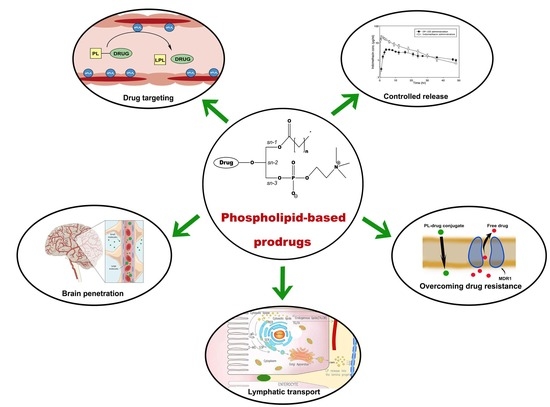
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
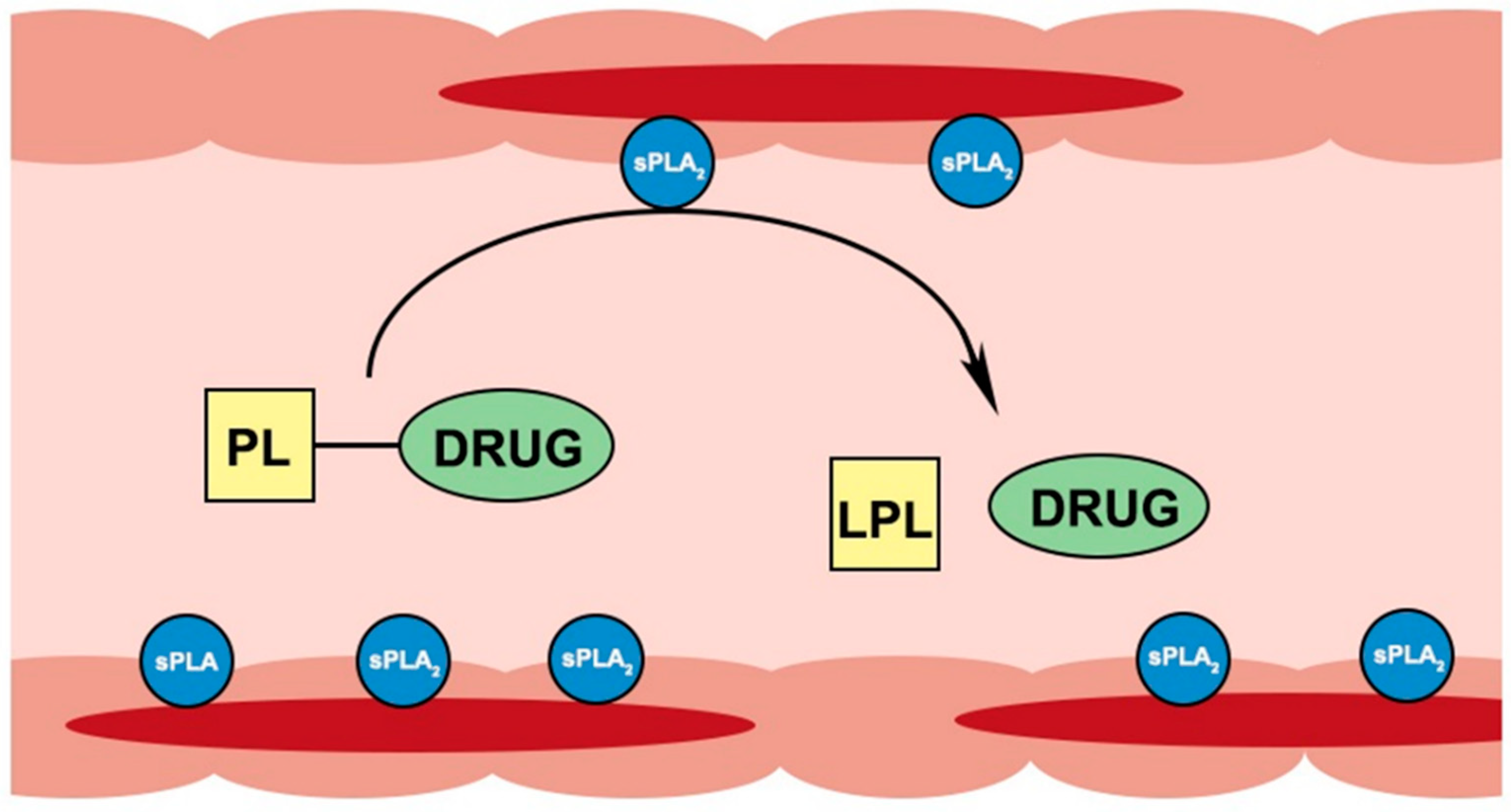
2. Processing Pathways of Phospholipids and Phospholipid-Based Prodrugs
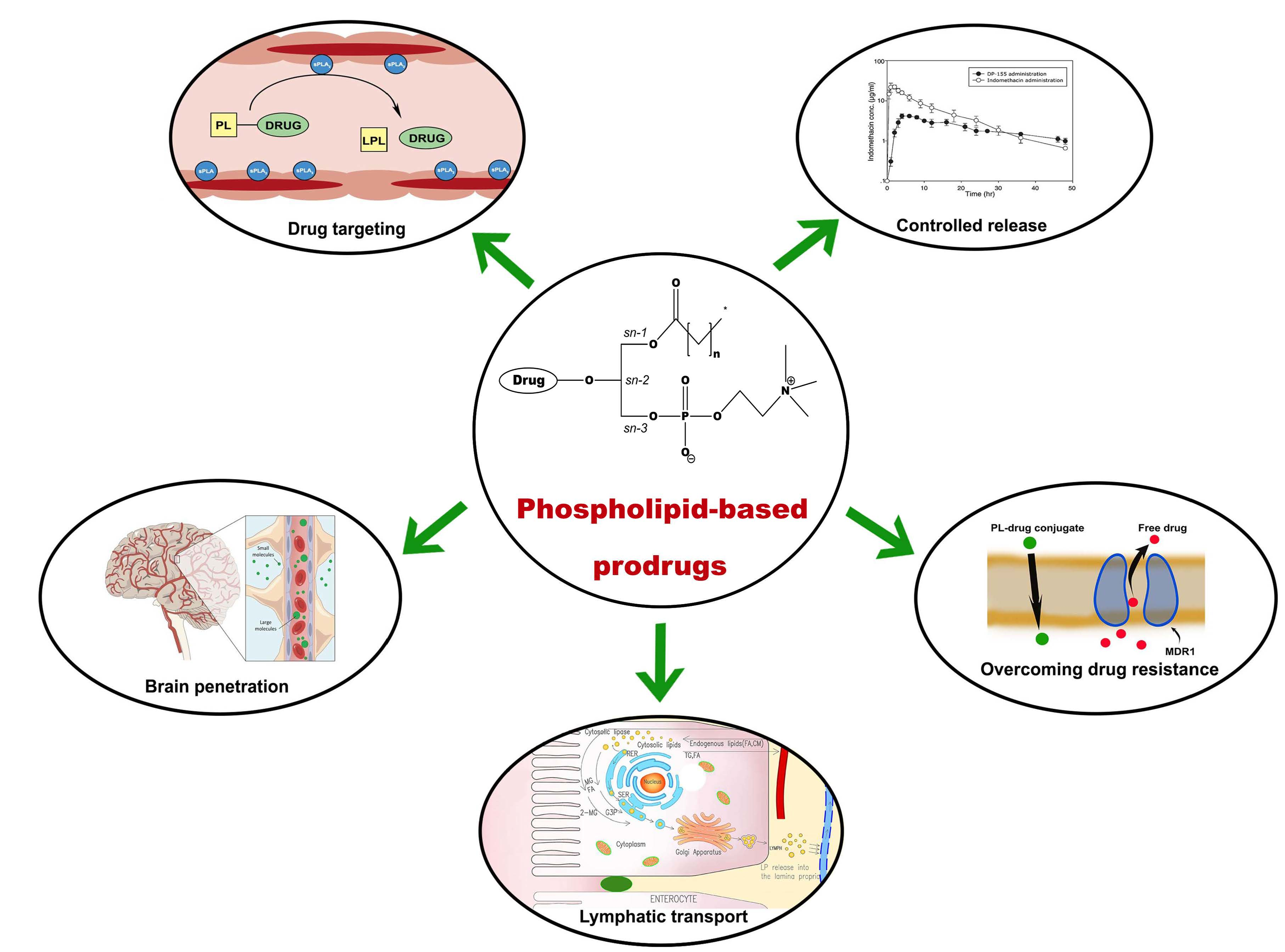
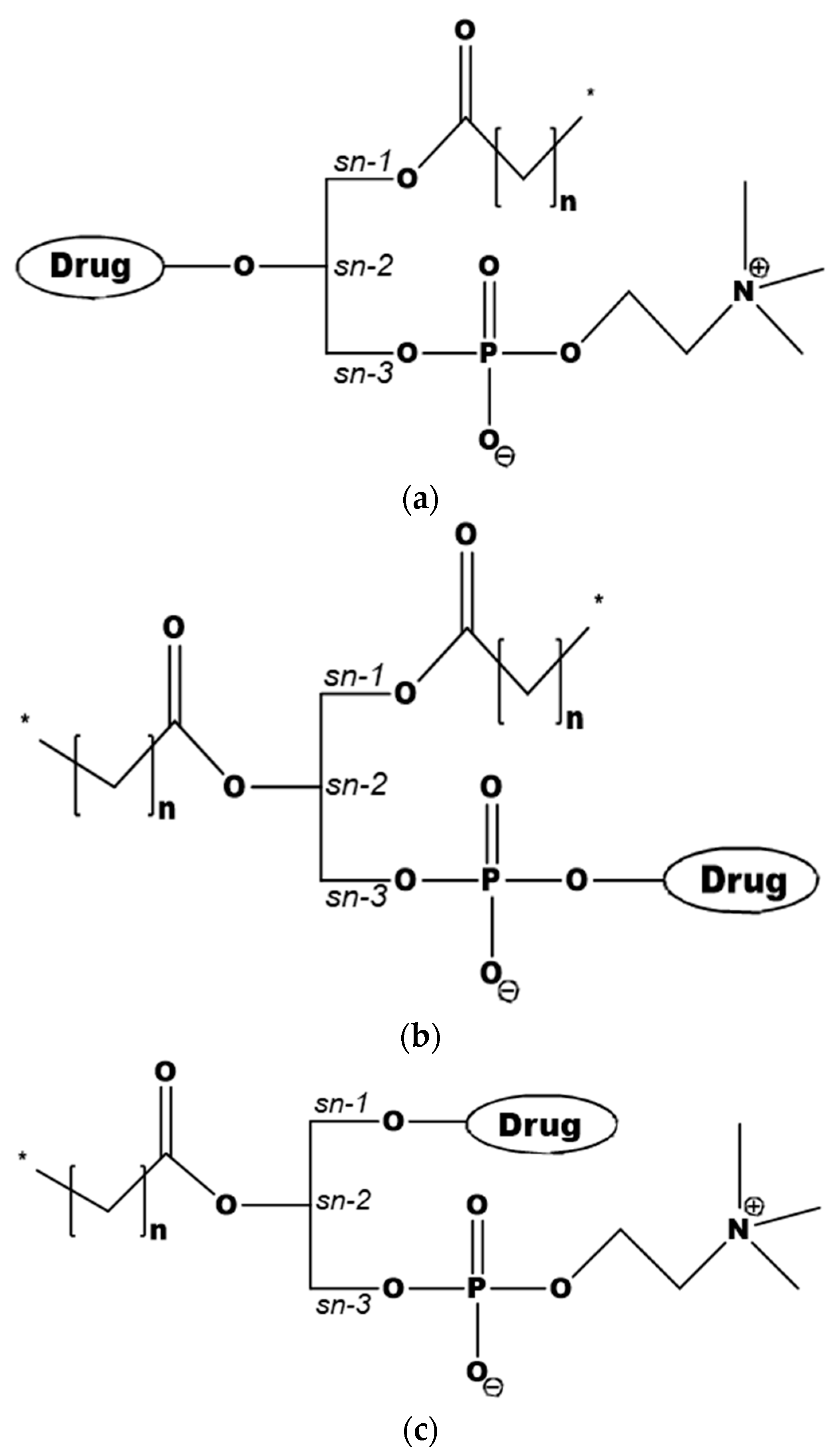
3. Phospholipid-Based Prodrugs: Structures and Applications
4. Computational Modeling of PL-Based Prodrugs
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stella, V.J. Prodrugs as therapeutics. Expert Opin. Ther. Patents 2004, 14, 277–280. [Google Scholar] [CrossRef]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Leesman, G.D.; Elliott, R.L. Improving intestinal absorption of water-insoluble compounds: A membrane metabolism strategy. J. Pharm. Sci. 1980, 69, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M. Rationale and applications of lipids as prodrug carriers. Eur. J. Pharm. Sci. 2000, 11, S15–S27. [Google Scholar] [CrossRef]
- Irby, D.; Du, C.; Li, F. Lipid–Drug Conjugate for Enhancing Drug Delivery. Mol. Pharm. 2017, 14, 1325–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Lipidic prodrug approach for improved oral drug delivery and therapy. Med. Res. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Khamis, M.; Agbaria, R.; Karaman, R. Targeted prodrugs in oral drug delivery: The modern molecular biopharmaceutical approach. Expert Opin. Drug Deliv. 2012, 9, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Zimmermann, E.M.; Ben-Shabat, S. Modern prodrug design for targeted oral drug delivery. Molecules 2014, 19, 16489–16505. [Google Scholar] [CrossRef] [PubMed]
- Han, H.K.; Amidon, G.L. Targeted prodrug design to optimize drug delivery. AAPS Pharm. Sci. 2000, 2, E6. [Google Scholar] [CrossRef]
- Sun, K.; Xu, H.; Hilfinger, J.L.; Lee, K.D.; Provoda, C.J.; Sabit, H.; Amidon, G.L. Improved Protease-Targeting and Biopharmaceutical Properties of Novel Prodrugs of Ganciclovir. Mol. Pharm. 2018, 15, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Amidon, G.L. The feasibility of enzyme targeted activation for amino acid/dipeptide monoester prodrugs of floxuridine; cathepsin D as a potential targeted enzyme. Molecules 2012, 17, 3672–3689. [Google Scholar] [CrossRef] [PubMed]
- Arouri, A.; Hansen, A.H.; Rasmussen, T.E.; Mouritsen, O.G. Lipases, liposomes and lipid-prodrugs. Curr. Opin. Colloid Interface Sci. 2013, 18, 419–431. [Google Scholar] [CrossRef]
- Haapamaki, M.M.; Gronroos, J.M.; Nurmi, H.; Alanen, K.; Kallajoki, M.; Nevalainen, T.J. Gene expression of group II phospholipase A2 in intestine in ulcerative colitis. Gut 1997, 40, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Haapamaki, M.M.; Gronroos, J.M.; Nurmi, H.; Irjala, K.; Alanen, K.A.; Nevalainen, T.J. Phospholipase A2 in serum and colonic mucosa in ulcerative colitis. Scand. J. Clin. Lab. Invest. 1999, 59, 279–287. [Google Scholar] [PubMed]
- Kennedy, B.P.; Soravia, C.; Moffat, J.; Xia, L.; Hiruki, T.; Collins, S.; Gallinger, S.; Bapat, B. Overexpression of the nonpancreatic secretory group II PLA2 messenger RNA and protein in colorectal adenomas from familial adenomatous polyposis patients. Cancer Res. 1998, 58, 500–503. [Google Scholar] [PubMed]
- Laye, J.P.; Gill, J.H. Phospholipase A2 expression in tumours: A target for therapeutic intervention? Drug Discov. Today 2003, 8, 710–716. [Google Scholar] [CrossRef]
- Minami, T.; Tojo, H.; Shinomura, Y.; Komatsubara, T.; Matsuzawa, Y.; Okamoto, M. Elevation of phospholipase A2 protein in sera of patients with Crohn's disease and ulcerative colitis. Am. J. Gastroenterol. 1993, 88, 1076–1080. [Google Scholar] [PubMed]
- Charman, W.N.; Porter, C.J.H. Lipophilic prodrugs designed for intestinal lymphatic transport. Adv. Drug Deliv. Rev. 1996, 19, 149–169. [Google Scholar] [CrossRef]
- Porter, C.J.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.; Charman, W.N. Intestinal lymphatic drug transport: An update. Adv. Drug Deliv. Rev. 2001, 50, 61–80. [Google Scholar] [CrossRef]
- Tauber, U.; Schroder, K.; Dusterberg, B.; Matthes, H. Absolute bioavailability of testosterone after oral administration of testosterone-undecanoate and testosterone. Eur. J. Drug Metab. Pharmacokinet. 1986, 11, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, N.L.; Charman, W.N.; Porter, C.J. Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Shackleford, D.M.; Porter, C.J.H.; Charman, W.N. Lymphatic Absorption of Orally Administered Prodrugs. In Prodrugs: Challenges and Rewards Part 1; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer: New York, NY, USA, 2007; pp. 653–682. [Google Scholar]
- Bernier-Latmani, J.; Petrova, T.V. Intestinal lymphatic vasculature: Structure, mechanisms and functions. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 510–526. [Google Scholar] [CrossRef] [PubMed]
- Sakai, A.; Mori, N.; Shuto, S.; Suzuki, T. Deacylation-reacylation cycle: A possible absorption mechanism for the novel lymphotropic antitumor agent dipalmitoylphosphatidylfluorouridine in rats. J. Pharm. Sci. 1993, 82, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Duvdevani, R.; Shapiro, I.; Elmann, A.; Finkelstein, E.; Hoffman, A. The oral absorption of phospholipid prodrugs: In vivo and in vitro mechanistic investigation of trafficking of a lecithin-valproic acid conjugate following oral administration. J. Control Release 2008, 126, 1–9. [Google Scholar] [CrossRef] [PubMed]
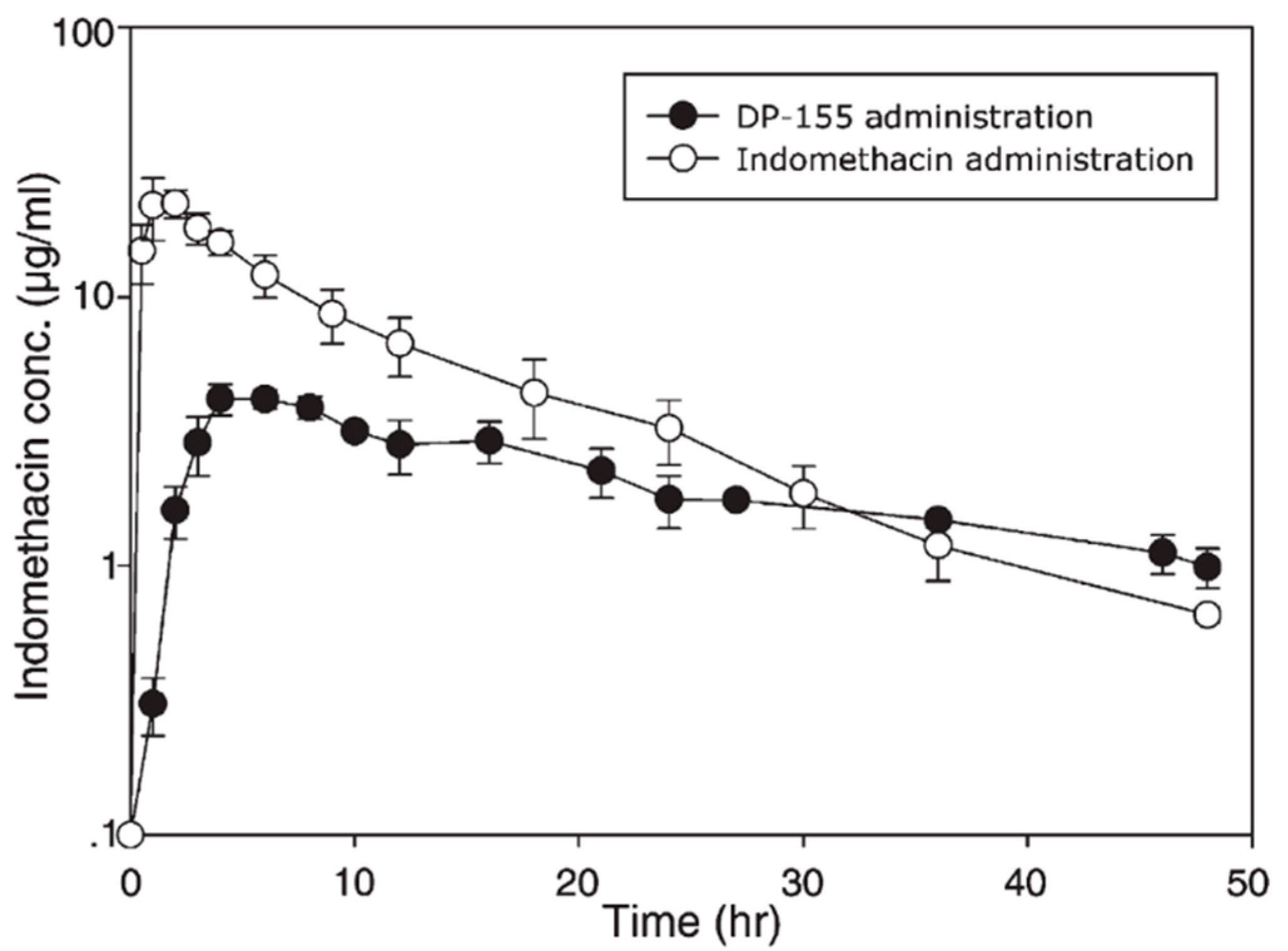
- Dahan, A.; Duvdevani, R.; Dvir, E.; Elmann, A.; Hoffman, A. A novel mechanism for oral controlled release of drugs by continuous degradation of a phospholipid prodrug along the intestine: In-vivo and in-vitro evaluation of an indomethacin-lecithin conjugate. J. Control Release 2007, 119, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Markovic, M.; Epstein, S.; Cohen, N.; Zimmermann, E.M.; Aponick, A.; Ben-Shabat, S. Phospholipid-drug conjugates as a novel oral drug targeting approach for the treatment of inflammatory bowel disease. Eur. J. Pharm. Sci. 2017, 108, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.S.; Andresen, T.L.; Davidsen, J.; Hoyrup, P.; Shnyder, S.D.; Bibby, M.C.; Gill, J.H.; Jorgensen, K. Secretory phospholipase A2 as a tumor-specific trigger for targeted delivery of a novel class of liposomal prodrug anticancer etherlipids. Mol. Cancer Ther. 2004, 3, 1451–1458. [Google Scholar] [PubMed]
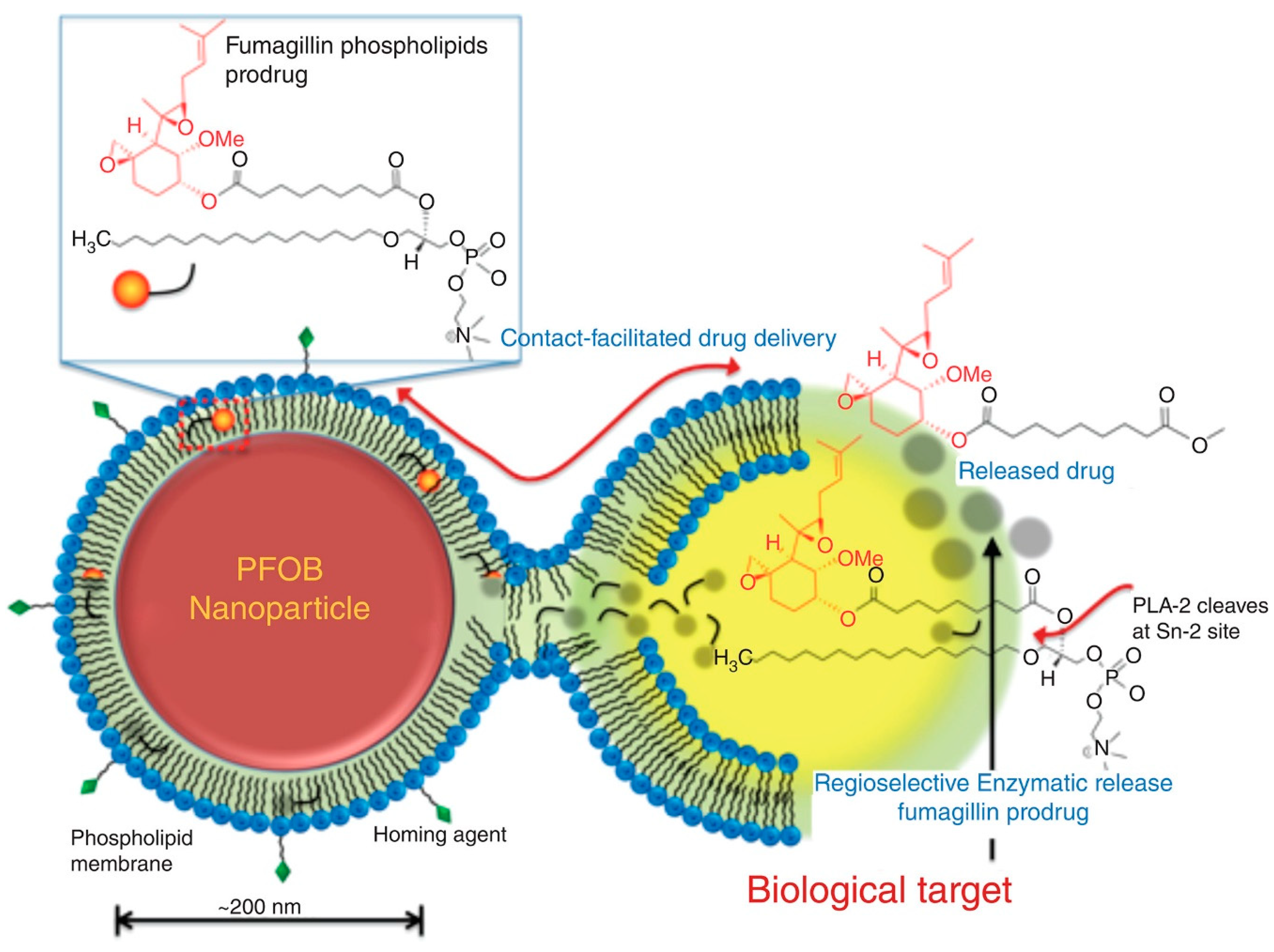
- Pan, D.; Sanyal, N.; Schmieder, A.H.; Senpan, A.; Kim, B.; Yang, X.; Hu, G.; Allen, J.S.; Gross, R.W.; Wickline, S.A.; et al. Antiangiogenic nanotherapy with lipase-labile Sn-2 fumagillin prodrug. Nanomedicine 2012, 7, 1507–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, R.L.; Greene, B.T.; Torti, S.V.; Kucera, G.L. A novel phospholipid gemcitabine conjugate is able to bypass three drug-resistance mechanisms. Cancer Chemother. Pharmacol. 2005, 56, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kucera, G.L.; Goff, C.L.; Iyer, N.; Morris-Natschke, S.; Ishaq, K.S.; Wyrick, S.D.; Fleming, R.A.; Kucera, L.S. Cellular metabolism in lymphocytes of a novel thioether-phospholipid-AZT conjugate with anti-HIV-1 activity. Antivir. Res. 2001, 50, 129–137. [Google Scholar] [CrossRef]
- Dvir, E.; Elman, A.; Simmons, D.; Shapiro, I.; Duvdevani, R.; Dahan, A.; Hoffman, A.; Friedman, J.E. DP-155, a lecithin derivative of indomethacin, is a novel nonsteroidal antiinflammatory drug for analgesia and Alzheimer's disease therapy. CNS Drug Rev. 2007, 13, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Pham, C.T.N.; Weilbaecher, K.N.; Tomasson, M.H.; Wickline, S.A.; Lanza, G.M. Contact-facilitated drug delivery with Sn2 lipase labile prodrugs optimize targeted lipid nanoparticle drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.J.; Adolph, S.K.; Subramanian, A.K.; Arouri, A.; Andresen, T.L.; Mouritsen, O.G.; Madsen, R.; Madsen, M.W.; Peters, G.H.; Clausen, M.H. Liposomal Formulation of Retinoids Designed for Enzyme Triggered Release. J. Med. Chem. 2010, 53, 3782–3792. [Google Scholar] [CrossRef] [PubMed]
- Rosseto, R.; Hajdu, J. Peptidophospholipids: Synthesis, phospholipase A2 catalyzed hydrolysis, and application to development of phospholipid prodrugs. Chem. Phys. Lipids 2014, 183, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, M.; Scriba, G.K. Drug-phospholipid conjugates as potential prodrugs: Synthesis, characterization, and degradation by pancreatic phospholipase A(2). Chem. Phys. Lipids 2000, 107, 143–157. [Google Scholar] [CrossRef]
- Isoherranen, N.; Yagen, B.; Bialer, M. New CNS-active drugs which are second-generation valproic acid: Can they lead to the development of a magic bullet? Curr. Opin. Neurol. 2003, 16, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Labiner, D.M. DP-VPA D-Pharm. Curr. Opin. Investig. Drugs 2002, 3, 921–923. [Google Scholar] [PubMed]
- Dahan, A.; Hoffman, A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur. J. Pharm. Sci. 2005, 24, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Hoffman, A. Use of a dynamic in vitro lipolysis model to rationalize oral formulation development for poor water soluble drugs: Correlation with in vivo data and the relationship to intra-enterocyte processes in rats. Pharm. Res. 2006, 23, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Tojo, H.; Shinomura, Y.; Matsuzawa, Y.; Okamoto, M. Increased group II phospholipase A2 in colonic mucosa of patients with Crohn’s disease and ulcerative colitis. Gut 1994, 35, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.W.; Dickey, W.D.; Saini, S.S.; Gourley, W.; Klimpel, G.R.; Chopra, A.K. Phospholipase A2 activating protein and idiopathic inflammatory bowel disease. Gut 1996, 39, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, I.; Edsfeldt, A.; Ko, N.Y.; Grufman, H.; Berg, K.; Bjorkbacka, H.; Nitulescu, M.; Persson, A.; Nilsson, M.; Prehn, C.; et al. Evidence supporting a key role of Lp-PLA2-generated lysophosphatidylcholine in human atherosclerotic plaque inflammation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Pruzanski, W.; Vadas, P.; Stefanski, E.; Urowitz, M.B. Phospholipase A2 activity in sera and synovial fluids in rheumatoid arthritis and osteoarthritis. Its possible role as a proinflammatory enzyme. J. Rheumatol. 1985, 12, 211–216. [Google Scholar] [PubMed]
- Tribler, L.; Jensen, L.T.; Jorgensen, K.; Brunner, N.; Gelb, M.H.; Nielsen, H.J.; Jensen, S.S. Increased expression and activity of group IIA and X secretory phospholipase A2 in peritumoral versus central colon carcinoma tissue. Anticancer Res. 2007, 27, 3179–3185. [Google Scholar] [PubMed]
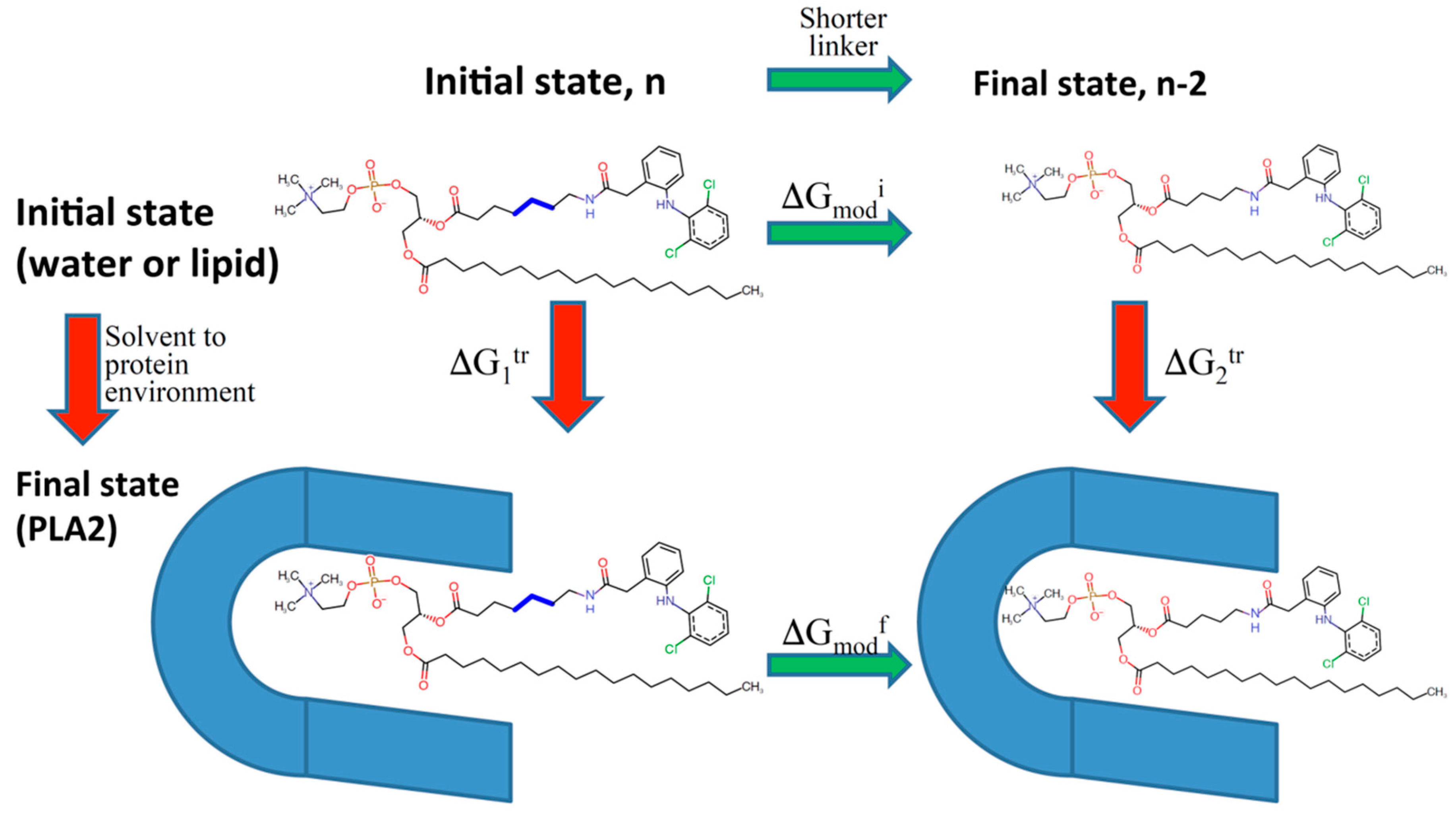
- Dahan, A.; Ben-Shabat, S.; Cohen, N.; Keinan, S.; Kurnikov, I.; Aponick, A.; Zimmermann, E.M. Phospholipid-Based Prodrugs for Drug Targeting in Inflammatory Bowel Disease: Computational Optimization and In-Vitro Correlation. Curr. Top. Med. Chem. 2016, 16, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Markovic, M.; Keinan, S.; Kurnikov, I.; Aponick, A.; Zimmermann, E.M.; Ben-Shabat, S. Computational modeling and in-vitro/in-silico correlation of phospholipid-based prodrugs for targeted drug delivery in inflammatory bowel disease. J. Comput. Aided Mol. Des. 2017, 31, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Rosseto, R.; Hajdu, J. Synthesis of oligo(ethylene glycol) substituted phosphatidylcholines: Secretory PLA2-targeted precursors of NSAID prodrugs. Chem. Phys. Lipids 2010, 163, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.F.; Yan, H.; Senpan, A.; Wickline, S.A.; Pan, D.; Lanza, G.M.; Pham, C.T. Suppression of inflammation in a mouse model of rheumatoid arthritis using targeted lipase-labile fumagillin prodrug nanoparticles. Biomaterials 2012, 33, 8632–8640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soodgupta, D.; Pan, D.; Cui, G.; Senpan, A.; Yang, X.; Lu, L.; Weilbaecher, K.N.; Prochownik, E.V.; Lanza, G.M.; Tomasson, M.H. Small Molecule MYC Inhibitor Conjugated to Integrin-Targeted Nanoparticles Extends Survival in a Mouse Model of Disseminated Multiple Myeloma. Mol. Cancer Ther. 2015, 14, 1286–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, G.M.; Jenkins, J.; Schmieder, A.H.; Moldobaeva, A.; Cui, G.; Zhang, H.; Yang, X.; Zhong, Q.; Keupp, J.; Sergin, I.; et al. Anti-angiogenic Nanotherapy Inhibits Airway Remodeling and Hyper-responsiveness of Dust Mite Triggered Asthma in the Brown Norway Rat. Theranostics 2017, 7, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; He, Y.; Zhang, S.; Qin, J.; Wang, J. Cell membrane-based nanoparticles: A new biomimetic platform for tumor diagnosis and treatment. Acta Pharm. Sin. B 2018, 8, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Luk, B.T.; Zhang, L. Cell membrane-camouflaged nanoparticles for drug delivery. J. Control Release 2015, 220, 600–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narain, A.; Asawa, S.; Chhabria, V.; Patil-Sen, Y. Cell membrane coated nanoparticles: Next-generation therapeutics. Nanomedicine 2017, 12, 2677–2692. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-M.J.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980. [Google Scholar] [CrossRef] [PubMed]
- Merkel, T.J.; Jones, S.W.; Herlihy, K.P.; Kersey, F.R.; Shields, A.R.; Napier, M.; Luft, J.C.; Wu, H.; Zamboni, W.C.; Wang, A.Z.; et al. Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proc. Natl. Acad. Sci. USA 2011, 108, 586. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.L.; Harada, T.; Christian, D.A.; Pantano, D.A.; Tsai, R.K.; Discher, D.E. Minimal “Self” Peptides That Inhibit Phagocytic Clearance and Enhance Delivery of Nanoparticles. Science 2013, 339, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Bu, L.-L.; Meng, Q.-F.; Cai, B.; Deng, W.-W.; Li, A.; Li, K.; Guo, S.-S.; Zhang, W.-F.; Liu, W.; et al. Antitumor Platelet-Mimicking Magnetic Nanoparticles. Adv. Funct. Mater. 2017, 27, 1604774. [Google Scholar] [CrossRef]
- Parodi, A.; Quattrocchi, N.; van de Ven, A.L.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Enzo, M.V.; et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2012, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhao, P.; Luo, Z.; Zheng, M.; Tian, H.; Gong, P.; Gao, G.; Pan, H.; Liu, L.; Ma, A.; et al. Cancer Cell Membrane–Biomimetic Nanoparticles for Homologous-Targeting Dual-Modal Imaging and Photothermal Therapy. ACS Nano 2016, 10, 10049–10057. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Lin, Z.; Jurado-Sánchez, B.; Lin, X.; Wu, Z.; He, Q. Stem Cell Membrane-Coated Nanogels for Highly Efficient In Vivo Tumor Targeted Drug Delivery. Small 2016, 12, 4056–4062. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Fang, R.H.; Thamphiwatana, S.; Luk, B.T.; Li, J.; Angsantikul, P.; Zhang, Q.; Hu, C.M.; Zhang, L. Modulating antibacterial immunity via bacterial membrane-coated nanoparticles. Nano Lett. 2015, 15, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Pei, Q.; Hu, X.; Zheng, X.; Liu, S.; Li, Y.; Jing, X.; Xie, Z. Light-Activatable Red Blood Cell Membrane-Camouflaged Dimeric Prodrug Nanoparticles for Synergistic Photodynamic/Chemotherapy. ACS Nano 2018, 12, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Alexander, P.; Kucera, G.; Pardee, T.S. Improving nucleoside analogs via lipid conjugation: Is fatter any better? Crit. Rev. Oncol. Hematol. 2016, 100, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaro, J.L. Lipid-based drug carriers for prodrugs to enhance drug delivery. AAPS J. 2015, 17, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.L.; Morris-Natschke, S.L.; Ishaq, K.S.; Fleming, R.A.; Kucera, G.L. Synthesis and Cytotoxic Activity of Two Novel 1-Dodecylthio-2-decyloxypropyl-3-phosphatidic Acid Conjugates with Gemcitabine and Cytosine Arabinoside. J. Med. Chem. 2003, 46, 4205–4208. [Google Scholar] [CrossRef] [PubMed]
- Pickin, K.A.; Alexander, R.L.; Morrow, C.S.; Morris-Natschke, S.L.; Ishaq, K.S.; Fleming, R.A.; Kucera, G.L. Phospholipid/deoxycytidine analogue prodrugs for the treatment of cancer. J. Drug Deliv. Sci. Technol. 2009, 19, 31–36. [Google Scholar] [CrossRef]
- Alexander, P.M.; Caudell, D.L.; Kucera, G.L.; Pladna, K.M.; Pardee, T.S. The novel phospholipid mimetic KPC34 is highly active against preclinical models of Philadelphia chromosome positive acute lymphoblastic leukemia. PLoS ONE 2017, 12, e0179798. [Google Scholar] [CrossRef] [PubMed]
- Brankatschk, M.; Eaton, S. Lipoprotein particles cross the blood-brain barrier in Drosophila. J. Neurosci. 2010, 30, 10441–10447. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.P.; Rodrigues, D.G.; Maranhao, R.C. Uptake of high density lipoprotein (HDL) cholesteryl esters by human acute leukemia cells. Leuk Res. 2005, 29, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Zensi, A.; Begley, D.; Pontikis, C.; Legros, C.; Mihoreanu, L.; Buchel, C.; Kreuter, J. Human serum albumin nanoparticles modified with apolipoprotein A-I cross the blood-brain barrier and enter the rodent brain. J. Drug Target 2010, 18, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Kucera, G.L.; Alexander, P.; Pladna, K.; Pardee, T.S. Abstract 4059: KPC34: A co-drug that combines a DNA damaging agent with a targeted therapy for the treatment of AML. Cancer Res. 2017, 77, 4059. [Google Scholar] [CrossRef]
- Andresen, T.L.; Jensen, S.S.; Madsen, R.; Jorgensen, K. Synthesis and biological activity of anticancer ether lipids that are specifically released by phospholipase A2 in tumor tissue. J. Med. Chem. 2005, 48, 7305–7314. [Google Scholar] [CrossRef] [PubMed]
- Andresen, T.L.; Davidsen, J.; Begtrup, M.; Mouritsen, O.G.; Jorgensen, K. Enzymatic release of antitumor ether lipids by specific phospholipase A2 activation of liposome-forming prodrugs. J. Med. Chem. 2004, 47, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Linderoth, L.; Fristrup, P.; Hansen, M.; Melander, F.; Madsen, R.; Andresen, T.L.; Peters, G.H. Mechanistic Study of the sPLA2-Mediated Hydrolysis of a Thio-ester Pro Anticancer Ether Lipid. J. Am. Chem. Soc. 2009, 131, 12193–12200. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/phospholipid-ether-drug-conjugate-clr-131?redirect=true (accessed on 9 October 2018).
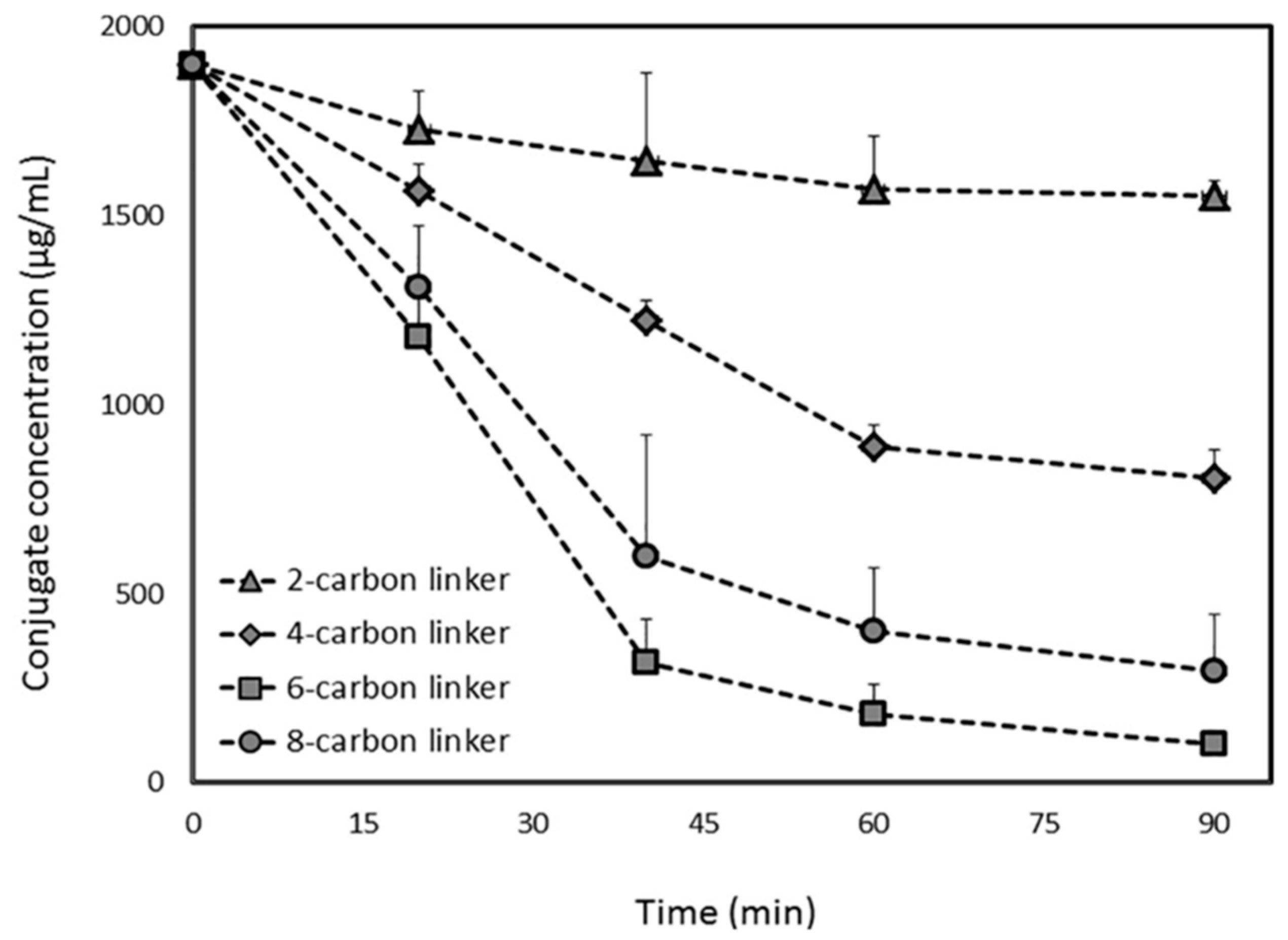
- Linderoth, L.; Andresen, T.L.; Jørgensen, K.; Madsen, R.; Peters, G.H. Molecular Basis of Phospholipase A(2) Activity toward Phospholipids with sn-1 Substitutions. Biophys. J. 2008, 94, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, H.; Fujii, S.; Tomoo, K.; Ishida, T.; Ikeda, K.; Tanabe, K.; Kitamura, K. Molecular dynamics simulation of 1,2-dilauroyl-L-phosphatidylethanolamine binding to phospholipase A2: An attempt to explain the selective hydrolysis of substrate fatty acid ester at position 2. J. Biochem. 1993, 114, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Goni, J.R.; Orozco, M.; Gelpi, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [PubMed]
- Arouri, A.; Mouritsen, O.G. Anticancer double lipid prodrugs: Liposomal preparation and characterization. J. Liposome Res. 2011, 21, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Arouri, A.; Mouritsen, O.G. Phospholipase A(2)-susceptible liposomes of anticancer double lipid-prodrugs. Eur. J. Pharm. Sci. 2012, 45, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Gliszczyńska, A.; Niezgoda, N.; Gładkowski, W.; Świtalska, M.; Wietrzyk, J. Isoprenoid-phospholipid conjugates as potential therapeutic agents: Synthesis, characterization and antiproliferative studies. PLoS ONE 2017, 12, e0172238. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S.; Ito, Y.; Hirokawa, T.; Hikiyama, E.; Yamada, S.; Shuto, S. Ligand-Phospholipid Conjugation: A Versatile Strategy for Developing Long-Acting Ligands That Bind to Membrane Proteins by Restricting the Subcellular Localization of the Ligand. J. Med. Chem. 2018, 61, 4020–4029. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Prospects and Challenges of Phospholipid-Based Prodrugs. Pharmaceutics 2018, 10, 210. https://doi.org/10.3390/pharmaceutics10040210
Markovic M, Ben-Shabat S, Keinan S, Aponick A, Zimmermann EM, Dahan A. Prospects and Challenges of Phospholipid-Based Prodrugs. Pharmaceutics. 2018; 10(4):210. https://doi.org/10.3390/pharmaceutics10040210
Chicago/Turabian StyleMarkovic, Milica, Shimon Ben-Shabat, Shahar Keinan, Aaron Aponick, Ellen M. Zimmermann, and Arik Dahan. 2018. "Prospects and Challenges of Phospholipid-Based Prodrugs" Pharmaceutics 10, no. 4: 210. https://doi.org/10.3390/pharmaceutics10040210
APA StyleMarkovic, M., Ben-Shabat, S., Keinan, S., Aponick, A., Zimmermann, E. M., & Dahan, A. (2018). Prospects and Challenges of Phospholipid-Based Prodrugs. Pharmaceutics, 10(4), 210. https://doi.org/10.3390/pharmaceutics10040210